Download presentation
Presentation is loading. Please wait.

1
中级物理化学 第五章 光化学 5.1 光和光化学技术基础 5.2 光化学基础 5.3 激发态的辐射跃迁与非辐射跃迁 5.4 激发态能量转移
第五章 光化学 5.1 光和光化学技术基础 5.2 光化学基础 5.3 激发态的辐射跃迁与非辐射跃迁 5.4 激发态能量转移 5.5 光致电子转移 5.6 光化学反应 5.7 光化学的应用

2
光和光化学技术基础 1. 光的基本性质 有关光性质的研究已经有300多年的历史,它和经典物理学和近代物理学的发展始终是密切相关的,研究证明,光具有二重性,即既可看成是显示波动性的电磁波,也可看成显示微粒性的光量子。

3
黑体辐射—能量量子化 理想黑体可吸收照射其上的全部能量,而与入射光波长无关。 基尔霍夫定律:一个好的吸收体也是一个好的发射体。
理想的黑体辐射源可由一大的球行腔做成,内壁是黑的,可吸收可见和不可见光区的全部辐射,辐射由腔壁上的一个小洞发射出来。 实验用黑体都是近似模拟的。一个可加热的炉子,炉腔内再放一个带小孔的耐热空心壳,炉子加热,空心壳通过小孔发射能量。辐射能量分布与炉温度有关。 经典物理的研究结果:W= σT4 , λmaxT=2.8978×10-3 米·开 能量量子化 :普朗克假设一个原子谐振子只能吸收或发射不连续的一份一份的能量,揭示了能量量子化,h=(6.6256±0.0005)×10-34焦耳·秒
×10-34焦耳·秒.")
4
光电效应 —光量子 赫兹发现光电子发射后,經典电磁理论无法解释:发射的光电子数与入射光强成比例,但光电子动能与入射光强无关,仅与入射光的频率成正比。 1905年爱因斯坦发展了普朗克的量子论,指出辐射场也是量子化的,它的吸收量也只能以量子hν(光子)来进行,并由此给出了光电效应方程。
来进行,并由此给出了光电效应方程。")
5
光压—光的粒子性特征 研究慧星时就提出了光辐射应当给被照物一定的压力—光压。
后经麦克斯韦、列别捷夫和盖拉赫等发展,指出光子不仅有能量也有动量,它是物质的一种形式。

6
偏振光 经典物理指出:由电偶极子振动所产生的光辐射是线偏振光或称平面偏振光,其中光的电场强度和符号随时间而改变,但电场的方向却不变。
实际光源的电矢量永远垂直于光的传播方向,但取向随时间是无规则变化的。自然光、太阳辐射、各种非相干辐射源所产生的光都是这样的。

7
电磁辐射

8
偏振光的产生 起偏过程:使光束产生某种行式的不对称性并选择某种偏振态。
起偏器:起偏器都是基于二向色性(或选择吸收)、反射、散射及双折射四种物理机制之一而产生起偏作用的。
、反射、散射及双折射四种物理机制之一而产生起偏作用的。")
9
2. 光学光谱区 由红外线经过可见光到紫外线这一频段将为光学光谱区,它仅是宽广的电磁波谱的一个小频段。

10
电磁波谱

11
光学光谱区各频段的性质 红外线频率范围3×1011Hz—4×1014Hz。任何物质都可以吸收和辐射红外线。
紫外线波段范围8× ×1017Hz (光子能量3.2ev-1.2×103ev)。光子能量与许多化学反应的能量在同一量级,大气中的臭氧吸收掉太阳的小于3000Å的紫外辐射。
。光子能量与许多化学反应的能量在同一量级,大气中的臭氧吸收掉太阳的小于3000Å的紫外辐射。")
12
电磁波谱其它频段光的性质 射频波段高频端用于电视和无线电广播。 微波由原子内层电子跃产生(30 cm-1mm)可以穿透大气。
高频段包括了X射线、γ射线等

13
光子能量单位 波数:每厘米长度内波的数目称波数。 电子伏特:一个电子伏特表示一个电子在一伏特电位降的场中所获能量。
1 ev=8066cm-1≈23kcal·m-1≈97kJ·mol-1 1光子/秒= / λ电子伏特,其中波长λ的单位为电子伏特。

14
3. 光源的作用和种类 光化学研究中光既是能量的来源,也是研究光化学反应动力学的信息源。 光源可分为两大类:非相干辐射源和相干辐射源。
3. 光源的作用和种类 光化学研究中光既是能量的来源,也是研究光化学反应动力学的信息源。 光源可分为两大类:非相干辐射源和相干辐射源。 非相干辐射源:黑体、太阳、白炽灯、普通的气体放电灯、脉冲闪光灯等。 相干辐射源:各类连续工作和脉冲工作的激光器。

15
常用非相干辐射源能谱分布 各种非相干光源所辐射的都不是单色光。 属热辐射类型的黑体、太阳、白炽灯都具有连续光谱。

16
太阳光

17
白炽灯 白炽钨丝灯所辐射的连续谱和黑体辐射相近,适合于产生可见光。
如要得到足够强度的紫外波段的光,其工作温度往往需要非常高,可在灯中加入少量的碘。

18
电弧灯 有些气体放电灯可提供能量基本集中在某几个置带或谱线区的光。主要有高压汞灯,低压汞灯,脉冲式高压氙灯(用于光解反应或作为激光的光泵)。
。")
19
高压氙灯

20
常用的激光器

21
激光器的特性 高单色性。 脉冲宽度可以很窄,适合于时间分辨 短脉冲可产生高峰值功率。 光束面积小,峰值光强可很高。 高方向性。 空间相干性。

22
市场上可以获得的光源

26
同步辐射光源 提供真空紫外光。 真空紫外光可以引发高能过程,包括高激发态和光电离过程。

27
单色辐射的获得 在光源的前面加各种滤光片、干涉滤光片、使用有光栅单色仪的分光仪器等。

28
光强的测量 太多数实验室采用相对的测量方法,这时用已知能量光谱分布的标准灯。
物理方法:用光辐射探测器,常用的有热探测器(如真空热电堆)、光电池、光电倍增管(光阳极表面具有选择吸收特性)。 化学方法:使用化学露光剂,常用草酸铁钾[ K3Fe(C2O4)3· 3H2O ]和草酸双氧铀。
、光电池、光电倍增管(光阳极表面具有选择吸收特性)。 化学方法:使用化学露光剂,常用草酸铁钾[ K3Fe(C2O4)3· 3H2O ]和草酸双氧铀。")
29
物理方法测光强的优缺点 热堆:测前后结点间产生的电热差并与温度对应。缺点为对室温有很大的敏感性。 光电池:容易带来噪声问题。
光电倍增管:适合弱光的检测(PMT)
")
30
4. 光化学反应、实验装置 光源发出的光经聚焦、滤光、穿过反应装置后由热堆、光电池等检测反应器中的吸光度。

31
激光扫描现场微区光电化学显微图像(In-situ PEM )测试系统示意图
测试系统示意图")
32
光化学中间体 光化学中间体包括:原子、自由基和离子等因光体而产生的碎片等物种;这些碎片的激发态;吸光物质产生的激发态以及它参与的荧光、磷光及无辐射跃迁等过程。

33
光化学中间体的鉴定与测定 光学光谱技术,特别在可见和紫外波段范围内通常是检测中间体的最灵敏和最有效方法之一。
发光、吸收、激光拉曼光谱、光电离方法、磁共振技术。

34
时间分辨和高分辨光化学 时间分辨技术是由20世纪50年代Norrish和Porter建立闪光解开始。

35
5.2 光化学基础 1. 分子轨道 分子轨道的组成 分子轨道是由构成分子的原子价壳层的原子轨道线性组合形成的。
5.2 光化学基础 1. 分子轨道 分子轨道的组成 分子轨道是由构成分子的原子价壳层的原子轨道线性组合形成的。 分子轨道法是一种用来描述分子中价电子的组合或分布的近似方法。

36
分子轨道能级示意图

37
分子轨道理论

38
分子轨道的类型 光化学涉及到五种类型的分子轨道: 未成键电子n轨道,成键电子π和σ 轨道,反键电子π*和σ*轨道。

39
n轨道 含有杂原子的分子中,杂原子的未共用电子在未成键轨道中,这种轨道不参与分子的成键体系。例羰基化合物中氧原子的未成键2P轨道。

40
π轨道和π*轨道 原子的2P轨道边靠边(平行)重叠形成 π 轨道。可表示为P轨道的线性组合,在分子平向上有一个节面。π 键电子在分子平面两侧对称分布。
重叠形成 π 轨道。可表示为P轨道的线性组合,在分子平向上有一个节面。π 键电子在分子平面两侧对称分布。")
41
σ轨道和σ*轨道 σ轨道是组成分子骨架的轨道。σ键比π键强。两个S、一个S和P或两个P轨道交盖都可形成σ键。

42
分子的基态电子组态 将电子填充到分子轨道上可得到分子的电子组态,如甲醛分子的基态可表示为:S0=(1S0)2 (1Sc)2(2S0)2(σCH)2(σCH’)2(σCO)2 (πCO)2 (n0)2 (π*CO)0 (σ*CO)0 。 与反应有关的是最高占有轨道和最低空轨道S0=(πCO)2(n0)2(π*CO)0。 分子激发态的电子组态则为电子跃迁到能量更高的轨道上。
2(n0)2(π*CO)0。 分子激发态的电子组态则为电子跃迁到能量更高的轨道上。")
43
分子的电子激发态组态 光化学中电子激发态是指将一个电子由低能轨道转移到高能轨道所形成的状态。 激发态 电子跃迁 电子组态
激发态 电子跃迁 电子组态 n, π* n→π* (πCO)2 (n0)1(π*CO)1 (σ*CO )0 n,σ* n→σ* (πCO)2(n0)1(π*CO)0 (σ * CO)1 π,π* π→π* (πCO)2(n0)2(π*CO)1 (σ * CO)1
2 (n0)1(π*CO)1 (σ*CO )0. n,σ* n→σ* (πCO)2(n0)1(π*CO)0 (σ * CO)1. π,π* π→π* (πCO)2(n0)2(π*CO)1 (σ * CO)1.")
44
甲醛的分子轨道示意图

45
激发态的多重态(多线态) 分子或原子的多重态是在强度适当的磁场影响下化合物在原子吸收和发射光谱中谱线的数目。谱线数为(2S+1),S为体系内电子自旋量子数的代数和,一个电子的自旋量子数可以是+1/2或-1/2。 PAULI不相容原理,同一轨道中的两个电子,必须是自旋配对的。在分子轨道上所有电子都是配对时2S+1=1,该状态称为单重态,用S表示。 分子中一个电子激发到能级较高的轨道上去,激发电子仍保持其自旋方向不变,S为0,体系处于激发单重态。自旋发生变化,S=1时,体系处于三重态,用T表示。 激发态的电子组态和多重态决定它的化学和物理性能。

46
羰基激发态的电子组态和多重度

47
激发态的能量 激发态的能量是决定它的化学和物理性能的另一个重要因素。
同一电子组态的激发态,单重激发态的能量比三重激发态的能量要高。能量差值取决于涉及轨道的重叠程度 有机光化学中,分子吸收光子后产生的电子激发态多为单线态。 一个分子的各种激发态的能量常用状态能级图来表示。

48
状态能级图

49
激发态的产生方法 有多种方法如放电、电离辐射、化学激活。 光化学中用的主要是分子吸收光产生激发态。

50
光的吸收 正常情况下,化合物的吸收特性用经验定律LAMBER-BEER定律描述,即光吸收可表示为:I=I0*10-εcl 或㏒(I0/I)=εcl ,其中I0为入射单色光强度, I为透射光强度,ε为消光系数,c为样品浓度,l为光程大小。 方程适用于强度不是很大的光。

51
电子跃迁选择定律 一个电子跃迁是允许的还是禁阻的,决定于跃迁过程中分子的几何形状和动量是否改变、电子的自旋是否改变、描述分子轨道的波函数是否对称以及轨道空间的重叠程度。 (A)Frank-Condon原理:在跃迁过程中,分子的几何形状和动量不变(跃迁时间10-15S)。 (B)自旋选择定则:电子跃迁过程中电子的自旋不能改变。
自旋选择定则:电子跃迁过程中电子的自旋不能改变。")
52
电子跃迁选择定律 (C)宇称禁阻:由跃迁所涉及的轨道的对称性所决定。(分子轨道的对称性取决于描述分子轨道的波函数在通过一个对称中心反演时符号是否改变。)对称G、反对称U、其中G→U或U→G的跃迁是允许的。 (D)轨道重叠:电子跃迁涉及的两个轨道在空间的同一个区域,即相互重叠时这种跃迁是允许的 。
轨道重叠:电子跃迁涉及的两个轨道在空间的同一个区域,即相互重叠时这种跃迁是允许的 。")
53
量子产率 量子产率是光子使用效率的一种量度。
原来的定义为体系吸收每个光子后所消耗的反应物分子数,由于该定义没有区分引发化学反应的原初光化学过程和次级反应,所以有时该值将大于1。 量子产率可区分为原初量子产率φ和总量子产率Φ。

54
量子产率 生产产物的物质的量或分子数 φ产物= --------------------------------
吸收辐射光的物质的量或光子数 产物的生成速率 = 所吸收辐射的强度

55
光化学反应的原初和次级过程

56
激发态寿命 激发态寿命有单分子寿命和辐射寿命两种定义。
单分子寿命( )指激发态分子的浓度衰减到原初浓度的1/e时所需要的时间。大多数有机化合物的S1态的寿命在10-9—10-6秒,T1态一般在10-3秒。 本征辐射寿命(0)可定义为在无辐射衰变过程存在时,受激分子的浓度衰减到起始浓度的1/e时所需要的时间。 = 0 , 为辐射过程的量子产率 。
指激发态分子的浓度衰减到原初浓度的1/e时所需要的时间。大多数有机化合物的S1态的寿命在10-9—10-6秒,T1态一般在10-3秒。 本征辐射寿命(0)可定义为在无辐射衰变过程存在时,受激分子的浓度衰减到起始浓度的1/e时所需要的时间。 = 0 , 为辐射过程的量子产率 。")
57
kasha规则 基态分子吸收光子后生成的不同激发态会很快失活到能量最低的激发态(10-13秒)
kasha规则:一切重要的光化学物理过程都是由最低激发单重态(S1)或最低激发三重态(T1)开始的。 激发态失活可经过的光化学和光物理过程包括: 辐射跃迁、无辐射跃迁、能量传递、电子转移和化学反应等。
或最低激发三重态(T1)开始的。 激发态失活可经过的光化学和光物理过程包括: 辐射跃迁、无辐射跃迁、能量传递、电子转移和化学反应等。")
58
辐射跃迁 分子由激发态回到基态或由高激到低激发态,同是发射一个光子的过程称为辐射跃迁,包括荧光和磷光。
荧光(Fluorescence):由多重度相同的状态间发生辐射跃迁产生的光,如S1→S0的跃迁。 磷光(Phosphorescence):不同多重度的状态间辐射跃迁的结果,如T1→S0;Tn→SO则较少。由于该过程是自旋禁阻的 ,因此与荧光相比其速度常数要小的多。
:由多重度相同的状态间发生辐射跃迁产生的光,如S1→S0的跃迁。 磷光(Phosphorescence):不同多重度的状态间辐射跃迁的结果,如T1→S0;Tn→SO则较少。由于该过程是自旋禁阻的 ,因此与荧光相比其速度常数要小的多。")
59
无辐射跃迁 激发态分子回到基态或高级激发态到达低激发态,但不发射光子的过程称为无辐射跃迁。
无辐射跃迁发生在不同电子态的等能的振动-转动能的之间,跃迁过程中分子的电子激发能变为较低的电子态的振动能,体系的总能量不变且不发射光子。 无辐射跃迁包括内转换和系间窜越。

60
内转换(IC)和系间窜越(ISC) 内转换(Internal Conversion):相同多重度的能态之间的一种无辐射跃迁,跃迁过程中电子的自旋不改变,如Sm~→Sn , Tm~→Tn ,时间10-12秒。 系间窜越(Intersystem Crossing):不同多重度的能态之间的一种无辐射跃迁。跃迁过程中一个电子的自旋反转,如S1~→T1或T1~→So。
:不同多重度的能态之间的一种无辐射跃迁。跃迁过程中一个电子的自旋反转,如S1~→T1或T1~→So。")
61
吸收、荧光和磷光发射过程的杰布朗斯基态图解

62
能量传递(ET) 一个激发态分子(给体D*)和一个基态分子(受体A)相互作用,结果给体回到基态,而受体变成激发态的过程。D*+A-D+A*,该过程中也要求电子自旋守恒,因此只有下述两种能量能递具有普遍性: 单重态—单重态能量传递:D*(S1)+A(SO)→D(SO)+A*(S1) 三重态—三重态能量传递:D*(T1)+A(SO)→D(SO)+A*(T1) 能量传递机制分为两种— 共振机制和电子交换机制。前者适用于单一单,后者两种传递都适用
+A(SO)→D(SO)+A*(S1) 三重态—三重态能量传递:D*(T1)+A(SO)→D(SO)+A*(T1) 能量传递机制分为两种— 共振机制和电子交换机制。前者适用于单一单,后者两种传递都适用.")
63
电子转移(ELT) 激发态分子可以作为电子给体,将一个电子给予一个基态分子,或者作为受体从一个基态分子得到一个电子,从而生成离子自由基对。D*+A→D+ + A A*+D→A- + D+ 基发态分子是很好的电子给体和受体。

64
化学反应 发生化学反应重生成基态产物,该过程是光化学研究的主要内容

65
Jablonski图解

66
5.3 激发态的辐射跃迁与非辐射跃迁 1. 吸收和辐射之间存在相关性
分子激发态的辐射跃迁是通过释放光子而从高能态失活到低能态的过程,是光吸收的逆过程。 辐射跃迁与光吸收之间有着密切的关系。

67
吸收和辐射跃迁都导致分子轨道电子云节面的改变
由于分子中电子的运动具有波动性,分子中电子运动轨道的能级是与其运动轨道的节面数相关的,其中,吸收光子的过程使分子的能量增加,导致相应分子轨道节面数增加,辐射过程使分子的能量降低,导致分子轨道节面数减少。

68
丁二烯的最高占有轨道和最低空轨道接面图

69
吸收和辐射遵从相同的选择规则 跃迁选择规则:跃迁是否容易发生主要与跃迁前后电子的自旋是否改变,跃迁涉及的分子轨道的对称性以及它们的重叠情况等因素有关。 化合物的摩尔消光吸收大体是从基态到激发态的跃迁容易与否的量度。( , m2/mol) 辐射跃迁遵从相同的选择规则,即电子自旋不发生改变、跃迁涉及的分子轨道对称性发生改变并有较大空间重叠时辐射跃迁容易发生。

70
吸收和辐射跃迁都将导致分子偶极矩的改变 从物理的角度上来说,跃迁矩(跃迁前后分子偶极矩的改变)是吸收光子的跃迁是否容易发生的量度,跃迁矩越大跃迁越容易发生。 辐射跃迁是电子从一个高能轨道回到低能分子轨道,因此分子中电子的排布也发生了改变,同样也要导致分子的偶极矩发生改变。
是吸收光子的跃迁是否容易发生的量度,跃迁矩越大跃迁越容易发生。 辐射跃迁是电子从一个高能轨道回到低能分子轨道,因此分子中电子的排布也发生了改变,同样也要导致分子的偶极矩发生改变。")
71
辐射跃迁与光子的吸收都遵从Franck—Condon原理
与分子的光吸收过程一样,辐射跃迁也是垂直跃迁,即在跃迁时分子的几何构型不发生变化,但此时由于辐射跃迁将产生一个“伸张”了的基态分子。

72
2.荧光的产生 吸收光谱和发射光谱 吸收光谱是将物质的吸光强度作为照射波长的函数所画出的图形。
发射光谱是将物质的发光强度(在某一固定吸收强度下)作为发射光谱波长的函数所画出的图形。 在多数情况下可以观察到发射光谱和吸收光谱有相同的形状,但对简单分子来说,由于其激发态构型和基态构型相差很大,二者之间不存在这种关系(称为Levschin规则)。
作为发射光谱波长的函数所画出的图形。 在多数情况下可以观察到发射光谱和吸收光谱有相同的形状,但对简单分子来说,由于其激发态构型和基态构型相差很大,二者之间不存在这种关系(称为Levschin规则)。")
73
荧光产生的条件 激发态是物质的高能和不稳定的状态,它可以通过无辐射跃迁、辐射跃迁或导致化合物分解等途径回到基态或其它低能状态。
一个化合物能够产生荧光的最基本条件是:它发生多重性不变的跃迁,并且在这个过程中所吸收的能量小于断裂最弱的化学键所需要的能量。[例:丁二烯的max=210nm,对应能量590kJ/mol,丁二烯最弱的化学键的键能为525 kJ/mol,因此不可能有荧光]

74
影响荧光的主要因素 荧光基团(Fluorophores) 荧光助色团(Fluorochromes) 增加稠合环可增强荧光
提高分子的刚性可增强荧光 激发态电子组态的影响 分子内的重原子将导致荧光量子产率的降低 增加溶剂的急性一般有利于荧光的产生 降低体系的温度可以提高荧光量子产率 其它如氢键、吸附、溶剂黏度增加等军可提高荧光的量子产率。

75
荧光基团 一个化合物要能发生荧光,在其结构中必须有荧光基团,即当下面这些基团是分子的共轭体系的一部分时,则该化合物可能产生荧光。

76
荧光助色团 可使化合物荧光增强的基团称为荧光助色团。
荧光助色团一般为给电子取代基,如-NH2和-OH等,相反吸电子基团如-COOH和-CN等将减弱或抑制荧光的产生。

77
增加稠合环可增强荧光 增加共平面的稠合环的数目,特别是当稠合环以线型排列时,将有利于体系内电子的流动,从而使体系发生跃迁所需要吸收的能量降低,有利于荧光的产生。

78
提高分子的刚性可增强荧光 刚性增加后,将减弱分子的振动,从而使分子的激发能不易因振动而以热能形式释放,同时分子刚性的增加常有利于增加分子的共平面性,从而有利于增大分子内电子的流动性,也有利于荧光的产生。

79
激发态电子组态的影响 根据Kasha规则,荧光通常都是从S1发出的。
有机化合物的第一激发单重态(S1)通常有两种电子组态,一是S1(π,π*),另一种是S1(n,π*),其中前者π到π*的跃迁是允许的过程而有利于荧光的产生,而后者是一禁阻过程则不利于荧光的产生。 如苯的S1态为前者,其荧光量子产率为0.2,二苯酮的S1态为后者其荧光量子产率为0。
通常有两种电子组态,一是S1(π,π*),另一种是S1(n,π*),其中前者π到π*的跃迁是允许的过程而有利于荧光的产生,而后者是一禁阻过程则不利于荧光的产生。 如苯的S1态为前者,其荧光量子产率为0.2,二苯酮的S1态为后者其荧光量子产率为0。")
80
分子内重原子的影响 分子内的重原子具有增强系间窜越的作用,将增大从S1态向T1态的窜越的速率常数和量子产率,从而使得分子内的重原子具有导致荧光量子产率增加的作用。

81
溶剂极性的影响 温度的影响 增加溶剂的极性,一般有利于荧光的产生。比如,喹啉、吡啶在非极性溶剂中无荧光,而在极性溶剂中它们都可以产生荧光。
降低体系温度可以提高荧光产率,这是因为,温度降低以后,分子的热振动降低,不利于以散热的方式失活,有利于其以释放光子的形式失活。

82
荧光强度、量子产率、速度常数和荧光寿命 荧光寿命、量子产率(φf)、速度常数和荧光强度是描述荧光性质的重要物理量。其中前三者都是一个化合物激发态的固有性质,荧光强度F则不是激发态的固有性质,它随物质所吸收的光强及激发光波长而改变。 荧光强度 F= φfIa =φfI0(1-e-2.303εcl)
")
83
荧光光谱和斯托克位移 荧光光谱:荧光强度是发射波长函数的图形通常被称为荧光光谱。
荧光发射是光吸收的逆过程,荧光光谱和吸收光谱有类似镜影的关系。 斯托克位移:荧光光谱较相应的吸收光谱红移,这被称为斯托克位移(Stoke’s shift)。 反斯托克位移:荧光光谱发生向短波方向的位移。
。 反斯托克位移:荧光光谱发生向短波方向的位移。")
84
蒽在溶液中的吸收和发射光谱

85
产生斯托克位移的原因 跃迁到激发态高振动能级的激发态分子,首先以更快的速率发生振动弛豫(1013秒-1数量级)而散失部分能量后到达零振动能级。 激发态分子的构型很快做进一步调整损失部分能量后达到较低能级的稳定态。 上述两种过程都导致荧光量子的能量低于被吸收光子的能量,从而使得与吸收光谱相比,荧光光谱发生向长波方向的位移。

86
高级激发态发射的荧光 有的化合物,如硫酮,它的S1与S0态之间的能隙很小,而S2和S1态间的能隙却较大,因此我们可观测到从S2态向S0态跃迁时所发射的荧光。

87
3. 磷光的产生 磷光是激发态分子失活到多重性不同的低能态时所释放出的辐射。
通常观测到的磷光都是从第一激发三重态(T1)向基态(S0)跃迁时所释放的辐射(Kasha规则)。 T1主要由S1态经系间窜越而形成,同时磷光发射过程是自旋禁阻过程,因此磷光的速率常数和强度都较荧光小。
向基态(S0)跃迁时所释放的辐射(Kasha规则)。 T1主要由S1态经系间窜越而形成,同时磷光发射过程是自旋禁阻过程,因此磷光的速率常数和强度都较荧光小。")
88
提高磷光量子产率的方法 重原子效应: 通常可以采用重原子效应、降低体系温度和向体系引入顺磁性分子等方法。
重原子效应包括分子内和分子外重原子效应。 分子内重原子效应指用Br和I等原子量大的原子取代氢原子;分子外重原子效应指用含有重原子的溶剂,如溴代丙烷、碘代丙烷等,代替普通轻溶剂。 重原子可增强旋轨偶合作用,从而提高电子自旋翻转的跃迁速率常数。

89
降低体系的温度 体系中引入顺磁性分子 降低温度后,与磷光过程竞争的无辐射跃迁速率可大大降低,从而提高磷光过程的量子产率。
比如,1-氯代萘在25℃时的量子产率小于10-4,而77K时则可达0.3。 体系中引入顺磁性分子 顺磁性分子(如O2和NO)有类似于重原子效应那样的增强旋轨偶合的作用,从而可以提高电子自旋翻转跃迁的速率和量子产率。
有类似于重原子效应那样的增强旋轨偶合的作用,从而可以提高电子自旋翻转跃迁的速率和量子产率。")
90
磷光光谱 以磷光发射强度作为波长函数表示的图形。 一个分子的磷光光谱总是在其荧光光谱的右侧---长波方向。
磷光光谱可以用于确定T1态的电子组态和能量。如T1态是(π,π*)态,则对重原子效应等表现敏感;从光谱峰位可以确定化合物的能量。 磷光一般较弱,通常在溶液中观测和研究一个化合物的磷光性质。
态,则对重原子效应等表现敏感;从光谱峰位可以确定化合物的能量。 磷光一般较弱,通常在溶液中观测和研究一个化合物的磷光性质。")
91
4. 延迟荧光 单重态的寿命一般为10-8秒,最长可达10-6秒,但有时却可以观察到单重态寿命长达10-3秒,这种长寿命的荧光被称为延迟荧光。 延迟荧光(delayed fluorescence)也被称为缓发荧光,它来源于从第一激发三重态(T1)重新生成的S1态的辐射跃迁。 S1 T1 S1 S0 +hυf 由于经历了上述过程,因此延迟荧光的寿命与三重态的寿命(~10-3秒)匹配,而远大于寻常的辐射寿命。
匹配,而远大于寻常的辐射寿命。")
92
产生延迟荧光的机理 根据过程,产生延迟荧光的机理可包括: 热活化延迟荧光(也称E型延迟荧光);
三重态—三重态湮灭延迟荧光(也称P型延迟荧光); 敏化延迟荧光。
; 敏化延迟荧光。")
93
热活化延迟荧光 其产生原因是,当第一激发单重态S1与第一激发三重态T1能差较小时,在高温体系中,T1态可从环境获取一定的能量后又达到能量更高的S1态(化合物的激发属于n π*跃迁时,能差通常小于10kcal/mol),新产生的S1态仍将以原有的量子产率发射荧光,这就是热活化延迟荧光。 热活化延迟荧光也被称为E型延迟荧光,这是由于这种现象受从四溴荧光素(eosin)上观测到。
上观测到。")
94
热活化延迟荧光过程图示

95
湮灭延迟荧光 当一个化合物的激发态的电子组态属( π,π*) 态时,S1与T1之间的能差一般较大,两个T1分子可通过靠近相互作用重新生成S1,然后生成荧光。 S1 T1 + T1 S1 + S0 S0 + S0 +hυp 这种现象最先从芘(pyrene)菲 (phenanthrence)的溶液中观察到,故得名P型延迟荧光。
菲 (phenanthrence)的溶液中观察到,故得名P型延迟荧光。")
96
湮灭延迟荧光过程

97
敏化延迟荧光 事实1:浓度为10-3mol/dm-3的菲溶液中加入10-7 mol/dm-3的蒽后可观察到由蒽产生的延迟荧光,而蒽在如此低的浓度下,由三重态—三重态湮灭形成能池的概率极低,可能的解释只能是菲起到了敏化传能的作用。 事实2:蒽萘混合体系的溶液可以产生较强的延迟荧光,而纯萘的溶液即使有很高的浓度也不能观察到它的延迟荧光。

98
敏化延迟荧光作用过程

99
敏化延迟荧光机理图解

100
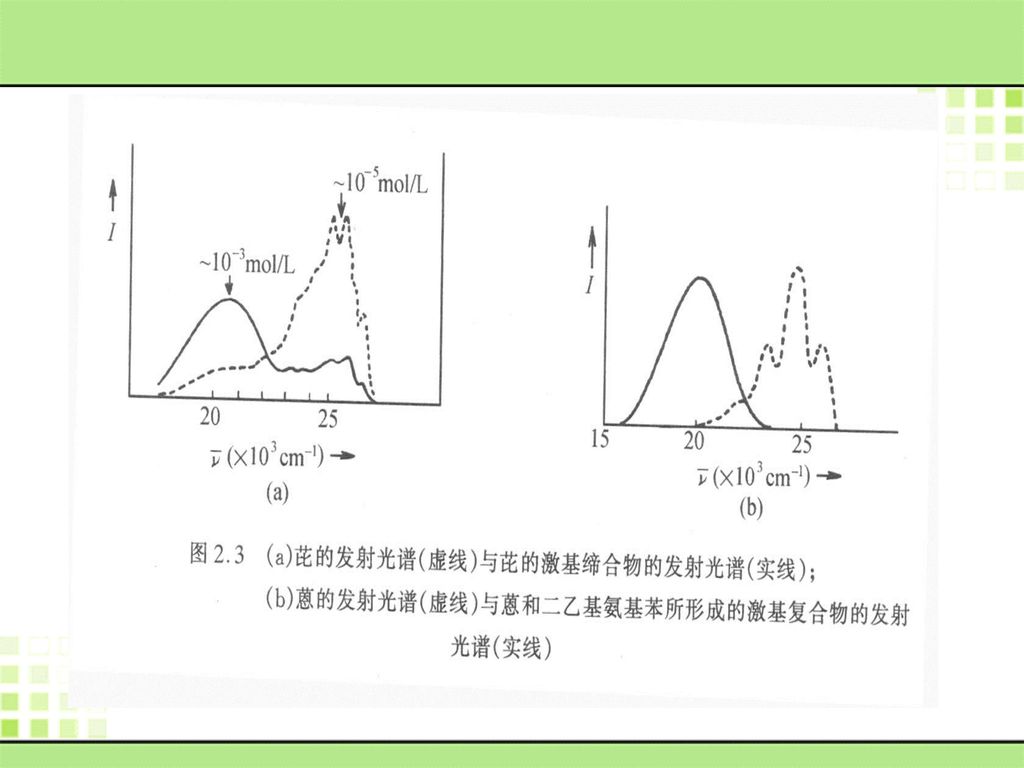
5. 激基缔合物和激基复合物 一个激发态分子以确定的化学计量与同种或不同种基态分子因电荷转移相互作用而形成的激发态碰撞络合物分别称为激基缔合物(excimer) 和激基复合物(exciplex)。 激基复合物也被称为激基络合物,二元激基缔合物和激基复合物可分别表示为:[AA]*和[AB]*。 它们不仅是一重要的光物理现象,也会影响其它物理过程,还是许多光化学反应的的中间体,对反应的发生、反应机理和产物结构的研究等都有重要的意义。
![5. 激基缔合物和激基复合物 一个激发态分子以确定的化学计量与同种或不同种基态分子因电荷转移相互作用而形成的激发态碰撞络合物分别称为激基缔合物(excimer) 和激基复合物(exciplex)。 激基复合物也被称为激基络合物,二元激基缔合物和激基复合物可分别表示为:[AA]*和[AB]*。](http://slidesplayer.com/slide/11131460/59/images/100/5.+%E6%BF%80%E5%9F%BA%E7%BC%94%E5%90%88%E7%89%A9%E5%92%8C%E6%BF%80%E5%9F%BA%E5%A4%8D%E5%90%88%E7%89%A9+%E4%B8%80%E4%B8%AA%E6%BF%80%E5%8F%91%E6%80%81%E5%88%86%E5%AD%90%E4%BB%A5%E7%A1%AE%E5%AE%9A%E7%9A%84%E5%8C%96%E5%AD%A6%E8%AE%A1%E9%87%8F%E4%B8%8E%E5%90%8C%E7%A7%8D%E6%88%96%E4%B8%8D%E5%90%8C%E7%A7%8D%E5%9F%BA%E6%80%81%E5%88%86%E5%AD%90%E5%9B%A0%E7%94%B5%E8%8D%B7%E8%BD%AC%E7%A7%BB%E7%9B%B8%E4%BA%92%E4%BD%9C%E7%94%A8%E8%80%8C%E5%BD%A2%E6%88%90%E7%9A%84%E6%BF%80%E5%8F%91%E6%80%81%E7%A2%B0%E6%92%9E%E7%BB%9C%E5%90%88%E7%89%A9%E5%88%86%E5%88%AB%E7%A7%B0%E4%B8%BA%E6%BF%80%E5%9F%BA%E7%BC%94%E5%90%88%E7%89%A9%EF%BC%88excimer%29+%E5%92%8C%E6%BF%80%E5%9F%BA%E5%A4%8D%E5%90%88%E7%89%A9%28exciplex%29%E3%80%82+%E6%BF%80%E5%9F%BA%E5%A4%8D%E5%90%88%E7%89%A9%E4%B9%9F%E8%A2%AB%E7%A7%B0%E4%B8%BA%E6%BF%80%E5%9F%BA%E7%BB%9C%E5%90%88%E7%89%A9%EF%BC%8C%E4%BA%8C%E5%85%83%E6%BF%80%E5%9F%BA%E7%BC%94%E5%90%88%E7%89%A9%E5%92%8C%E6%BF%80%E5%9F%BA%E5%A4%8D%E5%90%88%E7%89%A9%E5%8F%AF%E5%88%86%E5%88%AB%E8%A1%A8%E7%A4%BA%E4%B8%BA%EF%BC%9A%5BAA%5D%2A%E5%92%8C%5BAB%5D%2A%E3%80%82.jpg "它们不仅是一重要的光物理现象,也会影响其它物理过程,还是许多光化学反应的的中间体,对反应的发生、反应机理和产物结构的研究等都有重要的意义。")
101
激基缔合物 和激基复合物的性质 1、激基缔合物或激基复合物形成时,其发射光谱将展现出一个新的、强而宽的、长波、无结构发射峰。
2、激基缔合物的极性较弱,其发射光谱对溶剂的依赖性也较小;激基复合物的极性较强,其发射光谱对溶剂的依赖性也较大。 3、这种络合物的短寿命和不确定的振动特性导致其发射光谱没有振动结构,同时它也较单个激发态分子更稳定(能量更低),故其发射蜂总是在波长更长的位置。
,故其发射蜂总是在波长更长的位置。")
103
形成激基缔合物或激基复合物的条件 1、分子具有平面性,相互间距离达到~3.5×10-10米。 2、在溶液中分子有足够的浓度。
3、分子间的相互作用是吸引的。

104
激基缔合物或激基复合物的形式 1、可以是二元络合物,也可以是一个激发态分子与两个基态分子生成三元激基缔合物或激基复合物。

105
2、可以是同一个分子也可以生成分子内激基缔合物或激基络合物。其中,当分子内激基缔合物或激基络合物分子表示为A(CH2)nX时, n=3对这种电荷转移络合物的形成最有利。
nX时, n=3对这种电荷转移络合物的形成最有利。")
106
分子内激基缔合物的发射光谱

107
6. 无辐射跃迁的过程 通常,高振动激发态分子在能量的衰减过程中,首先通过振动驰豫并向环境散失一部分热能而达到激发态的零振动能级,然后再通过无辐射跃迁失活到能量更低的状态。 无辐射跃迁通常包括两个步骤:首先是在等能点上的跃迁----从激发态的零振动能级跃迁到低能状态(激发态或基态)的高振动能级;进而再经过振动驰豫失去过量的振动能达到零振动能级。
的高振动能级;进而再经过振动驰豫失去过量的振动能达到零振动能级。")
108
影响无辐射跃迁的因素 1、Franck--Condon积分。S1态与T1或S0态的核构型越相近,即Franck--Condon重叠积分越大,跃迁越容易发生。 2、能态密度。在始态或终态能量上,每单位能量间隔中的振动能级数称之为能态密度。对激发态分子来说,能态密度越大,则始态的零振动能级与终态的某一振动能级处于简并态的机会越多,也越有利于无辐射跃迁。

109
3、能隙。能隙是不同两个电子态的能差,能隙越小,两个不同电子态越容易发生共振,从而也越容易实现无辐射跃迁。
4、无辐射跃迁的选律与辐射跃迁相反。无辐射跃迁没有光子的吸收和发射,不要求电子云节面数发生变化,即始态与终态的分子轨道对称性不发生改变的无辐射跃迁是允许的。

110
7. 内转换的种类和特点 内转换包括Sn Sn-1和Tn Tn-1两类。
从S1 S0跃迁的内转换速率常数则低的多,一般在108s-1 数量级。 描述内转换的重要物理量是内转换速率常数和内转换量子产率。

111
影响内转换速率常数的因素 1、分子结构的影响。分子内的振动可以是无辐射跃迁的促进剂和受体,增加分子的刚性减小分子的振动将减小内转换。如叔丁基苯和甲苯的内转换速率常数分别为108s-1 和107s-1 。 2、能隙的影响。理论上内转换速率常数可达1013s-1,但实际却远远小于该值。两个相关状态的能隙是影响内转换速率常数的重要因素,并随能隙的增加呈指数下降。

112
3、重氢同位素的影响。原则上来说,有机物中的氢原子被重氢原子取代后,将使该分子的内转换速率常数降低。这是因为重氢原子使分子内振动减弱,从而不利于无辐射跃迁的发生。
4、温度的影响。温度升高将增加分子内的振动,因此可使内转换速率常数增加。 5、激发态电子组态。电子组态属(π,π*)态时速率常数较大,电子组态属于(n,π*)态时速率常数较小。
态时速率常数较大,电子组态属于(n,π*)态时速率常数较小。")
113
8. 系间窜越的形式 S1 Tn , Tn一般为T1。 T1 S0。

115
影响系间窜越的因素 1、温度 2、重原子 3、能隙 4、电子组态 5、氧的微扰作用 6、重氢的影响 8、能级错位的影响

116
5.4 激发态能量转移 1 . 激发态能量转移概述 能量转移是指能量从已经激发的粒子向未激发的粒子转移,或者在激发的粒子间转移的过程,这里指的粒子可以是原子、离子、基团或分子。 能量转移过程广泛存在于天然和人工合成体系中。分子激发能转移过程一般发生的距离范围约从1埃到100埃,时间从飞秒(10-15)到毫秒。 能量转移研究的目的在于:理解决定能量转移速率和效率的因素,并在此基础上实现对能量转移的控制和利用。
到毫秒。 能量转移研究的目的在于:理解决定能量转移速率和效率的因素,并在此基础上实现对能量转移的控制和利用。")
117
能量转移的分类 能量转移可发生在分子间和分子内。对分子间的能量转移来说,它既可以发生在不同的分子间,也可以发生在相同的分子间。分子内的能量转移则是指同一分子中的两个或几个发色团间的能量转移。 能量转移可以分为两大类:辐射转移和无辐射转移。

118
辐射能量转移的特点 辐射能量转移是一个两步骤过程: D* D + h υ h υ + A A *
辐射能量转移不涉及给体与受体间的直接相互作用,这种转移在稀溶液中可占主导地位。 转移的几率与激发态给体D* 发射的量子产率、受体A的浓度和吸收系数、以及D*的发射光谱与A的吸收光谱的重叠程度有关。 通过辐射机理发生的能量转移,给体发射寿命不改变,而且与介质的黏度无关。

119
无辐射能量转移的特点 无辐射能量转移是一个一步过程: D* + A D + A *
无辐射能量转移必须遵循体系总能量守恒定律,这要求D*D和AA *的能量相同。 自旋守恒与否是能量转移速率的重要决定因素。 无辐射能量转移过程是受不同机理支配的。这些机理包括:库仑转移机理、交换转移和通过键的超交换转移机理、激子转移机理。

120
2. 能量转移机理 库仑转移和交换转移机理 库仑转移机理---Förster理论。 交换转移---Dexter理论。

121
库仑转移与交换转移比较

122
激子转移机理 如果给体与受体相互作用大于单独分子内电子运动和核运动间的相互作用时,则称之为强相互作用或强偶合。这时给体与受体中振动子跃迁实际上是互相共振的,因此激发能转移速率比核振动快,而且核平衡位置在激发能转移时无实质性变化。这时的激发是离域的,也即在整个体系上分布,相当于用电子激发态定态来描述体系,这被定义为激子态,这种强相互作用引起的激发能转移也叫激子转移。

123
各能量转移机理的适用范围 实际问题研究中往往遇到多种机理存在于一体系中。影响转移机理最重要也是最直观的一个因素是给体与受体间的距离。一般来说,长距离转移属于库仑机理,接下来按距离由长至短,能量转移的机理依次是通过键的超交换机理、交换机理和激子机理。但这种距离界限并不是很明确的,所以确定一个体系中的能量转移的机理不是一件简单的事情。由单一因素来确定机理的做法往往是不可靠的。

124
3.能量转移研究方法 能量转移的实验研究方法: 1、稳态光谱法(常用的有吸收光谱、荧光发射和偏振光谱、线二色及圆二色谱等)
2、时间分辨光谱法(瞬态吸收和时间分辨荧光光谱) 能量转移的理论研究方法: 1、确定型方法 2、随机型方法
 能量转移的理论研究方法: 1、确定型方法. 2、随机型方法.")
125
一、概述 二、分子间的电荷(电子)转移 三、分子内的电荷转移 四、Marcus电子转移理论
转移 三、分子内的电荷转移 四、Marcus电子转移理论")
126
第五节 光致电子转移 1. 概述 电子转移图示 用分子轨道表示的一般电子转移过程,其中D为给体(donor) ,A为受体(acceptor)
 ,A为受体(acceptor)")
127
给体为激发态的光致电子转移 分子轨道表示的给体为激发态的光致电子转移过程

128
受体为激发态的光致电子转移 分子轨道表示的受体为激发态的光致电子转移过程

129
光致电子转移概念 光致电子转移是指电子给体或者电子受体首先受光激发,激发态的电子给体与电子受体之间或者电子给体与激发态的电子受体之间的电子转移反应。 与基态相比,激发态既是较好的电子给体,也是较好的电子受体,因此光致电子转移反应是光化学中较普遍的一类反应,同时电子转移也可以作为使激发态猝灭的一种重要途径。 光致电子转移反应包括初级电子转移反应和它的次级过程,前者指电子在激发态与基态分子之间通过转移形成电荷转移复合物的过程,后者指电子返回基态、系间窜越、离子对的分离或复合等过程。

130
激发态的猝灭和分子敏化 激发态分子和基态分子相互作用的过程中,处于激发态的能量给体D*回到基态,同时把能量转移到受体(基态)A分子上,使A从基态提升到某个激发态。其中,由于发生了能量转移,D*的发光过程减弱或完全停止,称为猝灭;A接受D*所给的能量后变为激发态,具有发射或激发态分子所应有的其它特性,这个过程称为敏化。 猝灭和敏化是同一过程的两个方面,就能量给体而言称为猝灭,以丧失部分或全部激发态的性质为标志,对能量受体而言称为敏化,它将表现出激发态的特征。
A分子上,使A从基态提升到某个激发态。其中,由于发生了能量转移,D*的发光过程减弱或完全停止,称为猝灭;A接受D*所给的能量后变为激发态,具有发射或激发态分子所应有的其它特性,这个过程称为敏化。 猝灭和敏化是同一过程的两个方面,就能量给体而言称为猝灭,以丧失部分或全部激发态的性质为标志,对能量受体而言称为敏化,它将表现出激发态的特征。")
131
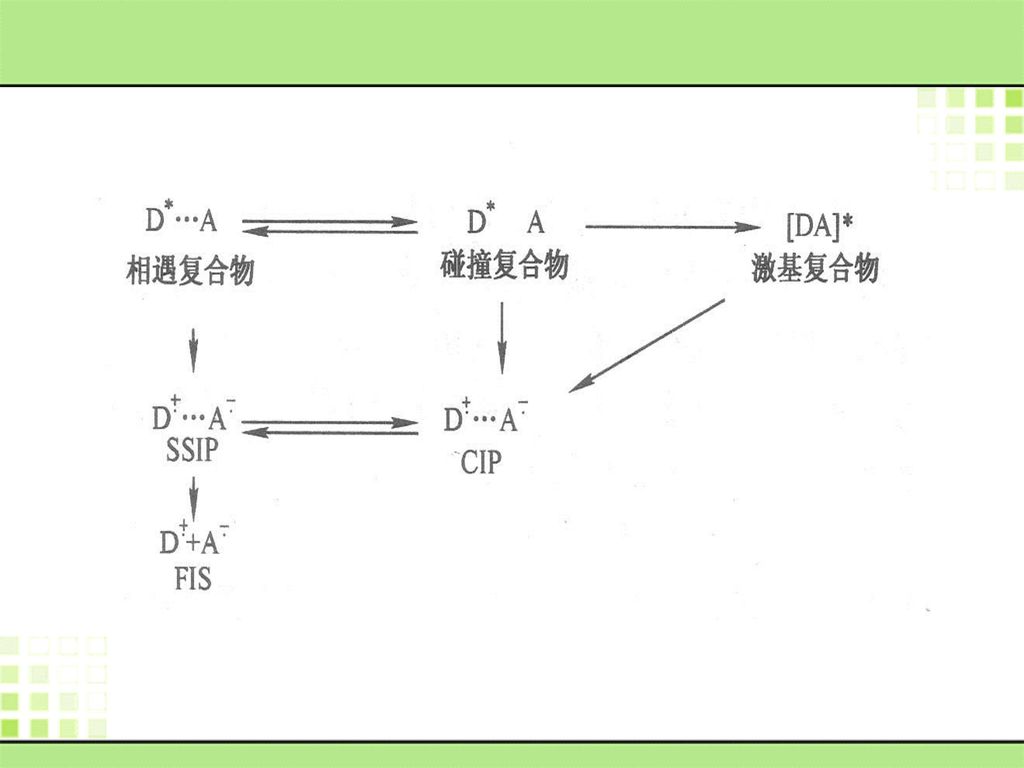
2. 分子间电荷转移的途径 在溶液中,独立存在的电子给体和电子受体的相互作用,由于溶剂的参与,可形成下列各种状态:相遇复合物(encounter complex)、碰撞复合物(collision complex)、激基复合物(exciplex)、接触离子对(contact ion pair,CIP)、溶剂分隔离子对solvent-separated ion pair,SSIP)、自由离子(free ions)、等。
、碰撞复合物(collision complex)、激基复合物(exciplex)、接触离子对(contact ion pair,CIP)、溶剂分隔离子对solvent-separated ion pair,SSIP)、自由离子(free ions)、等。")
132
相遇复合物中的电荷转移 相遇复合物是由激发态和基态分子相互作用生成的集合物。在溶剂笼中二者相距约0.7mm左右。激发态分子在衰变前与基态分子相遇并形成相遇复合物后,接着便发生碰撞、分离、再碰撞----。这其中,分子在形成相遇复合物期间可以完成激发态分子向基态分子的电荷转移过程,并有可能进而生成溶剂分隔的离子对。

133
碰撞复合物中的电荷转移 激基复合物中的电荷转移
激发态分子向基态受体间的电荷转移,如发生在碰撞复合物阶段,将立即形成紧邻离子对,也有可能生成溶剂分隔的离子对,并依溶剂极性的不同相互转化。 激基复合物中的电荷转移 形成激基复合物是一条重要的电荷转移途径。由于激基复合物的两部分都带有微量的电荷,因而具有较大的偶极矩。其中容易形成夹心结构的有机平面分子较容易形成激基复合物。

135
电子跳跃转移 给体与受体之间可以在溶剂参与下(并非必要条件)实现电子越过多个分子后的转移,这是一种长距离的电荷转移机制。 分子间电荷转移的研究方法 可以利用发射光谱、吸收光谱、闪光光解、CIDNP以及其它瞬态或时间分辨技术,但相对来说至今还没有形成一种十分有效的研究方法。

136
芘被DMA猝灭电荷转移机制

137
3. 非刚性分子内的电荷转移 非刚性分子具有构象变换位垒很低的特点,而且可接受温度、溶剂黏度和介电常数的影响。这种分子内的给体部分和受体部分的距离及所形成的激基复合物的构型、稳定性都具有可以变化的特点。

138
非刚性分子的分子内电荷转移过程(a为短链,b为长链)
")
139
刚性分子内的电荷转移 分子内电荷跳跃转移: 影响电荷转移的主要因素为给体和受体之间的相对取向和有效距离。
有机分子内的官能团之间可发生电荷转移

140
溶液中电子转移的途径

141
一、光化学反应的分类 二、光化学反应的特点 三、分子的光致断裂或光致离解 四、双键的异构化 五、光环合加成反应

142
5.6 光化学反应 1. 光化学反应的类型 光化学反应可以根据沿着反应坐标所经历的势能面的变化,分为绝对热的或非热的类型。其中反应发生在同一连续变化的势能面内,我们称这种反应是绝热的;若化学变化要交叉到另一个势能面,则称为非绝热的。 根据上述判据,在绝热的光化学反应中,反应物与产物,以至过渡态必须是相关的,产物处于激发态,可以借助荧光方法或光化学行为来检测。 在非绝热型反应中,如大多数的凝聚相光化学反应,受光激发后的分子体系会从能量高的势能面滑到低位,再经过无辐射跃迁回到基态后形成基态分子。

143
按势能面性质的光化学反应分类 A:绝热型; B:中间型; C:非绝热型

144
2. 激发态分子光化学反应的特点 通常基态分子的化学行为主要依赖于其最弱束缚电子的性质,而对处于激发态的分子来说,由于其内能和分子电子密度分布与基态分子完全不同,因此其化学性质与基态分子相比有很大的差异,表现出如下一些特点: 1、由于激发态分子核间的束缚能力常常比基态分子弱的多,因此易于离解,其中如果是被激发到排斥态而离解则其光离解效率可达1(光致离解)。 2、Franck—Condon原理,电子激发态的分子可能处于特定的振动和转动模式内发生反应,这在基态分子内通常是不可能的。
。 2、Franck—Condon原理,电子激发态的分子可能处于特定的振动和转动模式内发生反应,这在基态分子内通常是不可能的。")
145
3、通常分子内被激发的电子会到达很弱束缚的分子轨道内,因此分子具有很大的把电子转移给亲电子试剂的倾向(氧化)。
4、在无机化合物或络合物体系中,由于分子内或分子间的电荷转移会引起氧化还原反应。 5、一个体系中处于激发态的电子可以同另一个体系中未配对电子发生相互作用,以至形成新的化学键。

146
3. 光解离 当分子吸收的光子能量大于或等于分子的某化学键的离解能时,分子就会直接离解,光解离作为最基本的光化学过程,它可以导致处于电子激发态的分子发生光化学反应。 光解离有三种主要类型:光学解离、预解离和诱导解离。 在光解离过程中,产物分子的对称性必须与反应物分子的对称性相关,其中在绝热反应中反应分子和产物分子必须位于相同的势能面上。

147
气相光化学 一、碳氢化合物 1、烷烃在真空紫外区有很强的( *)允许跃迁,吸收系数很大(104)。甲烷的吸收从144nm开始,高级烷烃的吸收波长略有红移,在 nm。
允许跃迁,吸收系数很大(104)。甲烷的吸收从144nm开始,高级烷烃的吸收波长略有红移,在 nm。")
148
2、不饱和烃的最大吸收波长在180nm左右,属于 *跃迁。共轭体系增大后,吸收波长红移。不饱和烃的光化学反应包括异构化和光解离。

149
3、多烯烃的光解离只在低压气相中发生,加入外部惰性气体后可受到抑制。
4、简单的芳烃在近紫外区有中等的吸收强度。短波长的光可使苯发生完全解离,而长波长的光则只能使苯产生激发态,继而发生光化学反应式辐射失活。

150
二、羰基化合物 1、诺瑞什I型光解:在光作用下,羰基化合物的位置的光解反应。

151
2、诺瑞什II型光解:在光的作用下,在位置上有H的酮,先发生自身光还原,然后开裂称烯烃和烯醇,后者经异构化变为相应的酮。

152
溶液中的光化学 与气相光化学反应相比,在溶液中光的吸收和激发态的弛豫过程都要受到溶剂的影响,表现为溶质的能量发生变化,吸收光谱的强度也要受到溶剂的影响,吸收谱线的碰撞加宽,转动精细结构消失。 溶液中激发态的弛豫过程发生明显的变化的重要原因有:碰撞频率增加使得原初光化学过程的量子产率降低;激发态分子与溶剂分子间发生反应;激发态解离生成的碎片也可能和溶剂分子发生反应。 溶液中的光化学过程与气相光化学过程的差别,可以认为与溶质分子在溶液中处于溶剂笼中有关

153
离子型物种的光化学 离子型物种的光化学是溶液中的另一类型的光化学,和中性分子的不同主要表现为离子的原初光化学过程通常具有氧化还原的特征。(光解水溶液中的离子时,可以产生出电子) 多电子解离和电离 以高功率激光为代表的高能辐照下的多光子激励,和继而引发的光化学过程,已经成为光化学中一个十分重要的领域。 实现对分子的多光子激励,有两种常用的机制:一种是共振激励机制,通过分子n个光子的同步吸收,使其经共振激励而升至很高的电子束缚态(或连续的解离态或预解离态);另一种为非共振激励。即中间能级是实际存在或部分存在,分子的激励过程类似于在间隔基本相近的阶梯上的攀升过程。
;另一种为非共振激励。即中间能级是实际存在或部分存在,分子的激励过程类似于在间隔基本相近的阶梯上的攀升过程。")
154
4. 碳-碳双键的异构化反应 双键的异构化是由与双键相联的一端的基团相对于另一端的基团发生了180度变化引起的。可以通过热化学方法、催化方法和光化学方法实现。

155
烯烃的异构化 最简单的例子是反式(t)二氘代乙烯气相以147nm和148nm的光照射得到顺式异构体(c),其反应机理式通过激发单重态的“P”态,然后内部转化为基态单重态从而得到c或t。 由于烯烃的激发单重态和三重态之间的能隙太大,直接光照只能以激发单重态机理进行异构化,而烯烃的激发三重态的异构化只能通过光敏化的方法(常见 三重态敏化剂是羰基化合物,如丙酮、苯乙酮、二苯酮等)。
。")
157
氮-氮双键的异构化 偶氮苯类化合物是典型的含氮-氮双键的化合物。下图以偶氮苯为例给出其光异构化机理图示。

158
碳-氮双键的异构化 碳-氮双键的异构化机理和氮-氮双键的异构化类似

159
5. 环合加成反应 光环合加成反应是指在光的作用下,一组m个原子与另一组n个原子的分子进行反应,生成m+n个原子的环状化合物分子的反应。
5. 环合加成反应 光环合加成反应是指在光的作用下,一组m个原子与另一组n个原子的分子进行反应,生成m+n个原子的环状化合物分子的反应。 最常见的光环合加成反应是[2+2]、[4+4]和[1+2]类型;[2+4]和[3+2]类型则较少。 就光环合加成反应的过程来说,大致可以分为两类,一类是协同反应,另一类是分布反应。 根据反应物是相同或不同分子,光环合加成又可分为分子内环合加成和分子间环合加成。 光环合加成反应的研究主要集中在:碳-碳不饱和键之间的环合加成反应;羰基和硫羰基参与的光环合加成反应;含碳-氮不饱和键的光环合加成反应。

161
对苯二醌与苯乙烯的光环合加成反应

162
光环合加成图示

163
第七节 光化学的应用 概述 内容

164
光化学的应用前景 光化学与人们的日常生活密切相关,它涉及到环境、材料、信息和能源等方面,在二十一世纪优先发展的高新技术中占有重要的的位置。应用前景十分广泛。

165
内容 一、大气光化学 二、生物光化学 三、光化学信息储存 四、光致变色 五、高聚物的光化学 六、超分子光化学和光化学分子器件
七、半导体和纳米材料

166
1. 大气光化学 大气的起源与进化 大气污染与保护

167
1.1 大气的起源 不少证据表明,地球上原本并不存在大气。
在大气的形成过程中,地球上的火山喷发、太阳光的照射、生物体的出现起到了重要的作用。 在大气形成的最初阶段,由于火山喷发物中不含氧,因此最早的大气中必须以N2、CO2和H2O为主,同时有痕量的还原性气体,如H2和CO。 在生命出现之前, O2来源于短波紫外线对水的光解: H2O + hv 2H + O,然后再生成氧和氢分子。由于O2和CO2可以遮蔽紫外光,这使得O2的浓度上限受到限制,只有现在大气水平的千分之一。 光合作用是目前唯一知道的能够使氧的水平上升到现有水平的途径(时间跨度约需20亿年)。
。")
168
1.2 大气的进化 生命约在75亿年前出现在地球上,当时氧几乎为零,细菌的光合作用的氢源为H2S,随着蓝球藻的繁殖,65亿年前水成为光合作用的新氢源,而同时生成的氧被放到大气中。 早期的氧浓度有利于生命的有机前体的发展。 17亿年前氧浓度接近现有大气水平,并且在大气层上部出现臭氧层。其中,地球大气中的分子氧可以滤去太阳辐射中波长低于230nm的紫外部分, nm部分则由臭氧滤去。 O3的生成决定于生物产生的氧,也与生物产生的致使其破坏的痕量气体有关。 总体上可以说,在过去的年代里,生命的形成和生态学是与当时的大气组成相关的,同时生命机体的活动与变化对大气的组成和变化又产生重要的影响。

169
1.3 大气平流层 大气层可以分为临近地球的对流层(距地表15-20km)及其上面的平流层。臭氧主要存在于平流层中。
及其上面的平流层。臭氧主要存在于平流层中。")
170
1.4 臭氧层的形成理论 1930年,Chapman提出了形成臭氧层的基本过程(左)。其中下面过程中最后给出的反应可以可以被大气中的痕量物质(包括H、OH、NO、Cl等)所催化(右)。
。其中下面过程中最后给出的反应可以可以被大气中的痕量物质(包括H、OH、NO、Cl等)所催化(右)。")
171
1.5臭氧层的破坏 人类活动生成了大量的NOx,可破坏臭氧层。
氟氯烃(CFCs)的排放,如常用做溶剂及制冷剂的氟氯烃CF2Cl2和CFCl3,它们在化学上表现为惰性,在对流层中的寿命可达50年,一旦进入平流层将受到短波紫外光线的作用发生光解,从而产生原子Cl,产生的Cl参与致使臭氧层破坏的ClOx循环。
的排放,如常用做溶剂及制冷剂的氟氯烃CF2Cl2和CFCl3,它们在化学上表现为惰性,在对流层中的寿命可达50年,一旦进入平流层将受到短波紫外光线的作用发生光解,从而产生原子Cl,产生的Cl参与致使臭氧层破坏的ClOx循环。")
172
臭氧层小知识之一 什么臭氧层:臭氧是氧气的一种同素异形体(由相同的元素组成,但分子结构不同) 。顾名思义,臭氧又一种刺鼻的气味,所以得此恶名。在大气层的10公里到50公里高度的区域,臭氧有相当的浓度,叫做臭氧层。 臭氧层破坏的影响:臭氧层被大量损耗后,吸收紫外辐射的能力大大减弱,导致到达地球表面的紫外线明显增加,给人类健康和生态环境带来多方面的的危害,目前已受到人们普遍关注的主要有对人体健康、陆生植物、水生生态系统、生物化学循环、材料、以及对流层大气组成和空气质量等方面的影响。

173
臭氧层小知识之二 臭氧由3个氧原子结合而成,绝大部分存在于离地面25-30公里处的大气平流层,被称为臭氧层。臭氧层是法国科学家法布里20世纪初发现的,它能吸收99%以上对人类有害的太阳紫外线,为地球上的生物提供天然保护屏障,使其免遭紫外线的伤害,因而被称为地球保护伞。但是自20世纪70年代以来,臭氧层发生严重耗损,80年代中期科学家首先发现了南极上空的臭氧层空洞。1998年9月中旬到12月中旬,南极上空臭氧层的空洞达到2720万平方公里,是观测史上最大的臭氧层空洞,而且持续时间也最长。

174
臭氧层小知识之三 专家们认为,导致臭氧层空洞出现是人类大量使用氯氟烃化学制品(可用于冰箱、空调的制冷剂)引起的恶果。臭氧层破坏是当今全球环境问题之一。为保护臭氧层,国际社会于1985年签署了《保护臭氧层维也纳公约》,并于两年后制定了《关于消耗臭氧层物质的蒙特利尔议定书》,开始在全球范围内限制并逐步淘汰消耗臭氧层的化学物质。由于国际社会采取了切实措施,近年来释放到空气中的氯氟烃开始减少,大气中这种物质的总含量也于2000年达到顶峰后开始下降,南极洲上空的臭氧层空洞开始缩小。
引起的恶果。臭氧层破坏是当今全球环境问题之一。为保护臭氧层,国际社会于1985年签署了《保护臭氧层维也纳公约》,并于两年后制定了《关于消耗臭氧层物质的蒙特利尔议定书》,开始在全球范围内限制并逐步淘汰消耗臭氧层的化学物质。由于国际社会采取了切实措施,近年来释放到空气中的氯氟烃开始减少,大气中这种物质的总含量也于2000年达到顶峰后开始下降,南极洲上空的臭氧层空洞开始缩小。")
175
1.6 对流层中的光化学过程 与平流层中的化学过程主要由氧原子和臭氧主宰不同,对流层的化学过程是由氢氧自由基所主宰的。
对流层中最终要的光化学活性物种为O3、NO2、和HCHO,它们都能够间接产生OH,从而引发氧化链的形成。

176
1.7 光化学烟雾

177
2 生物光化学 光合作用。

178
3 光化学信息储存 光化学成像 光子型信息存储

179
3.1光化学成像及成像材料 用照相术将明暗的景象永久记录下来的方法,是光化学过程中应用最普遍也是最成功的过程之一。
光化学成像过程中要用到感光材料,现在已经得到应用或者有很大应用前景的感光材料有:银盐感光材料、非银盐感光材料系列(如重氮成像材料、自由基成像体系感光材料)。
。")
180
3.2 重氮成像材料 重氮成像材料是一种开发较早、应用也较广泛的非银盐感光材料,广泛应用于缩微、复制和印刷等领域。
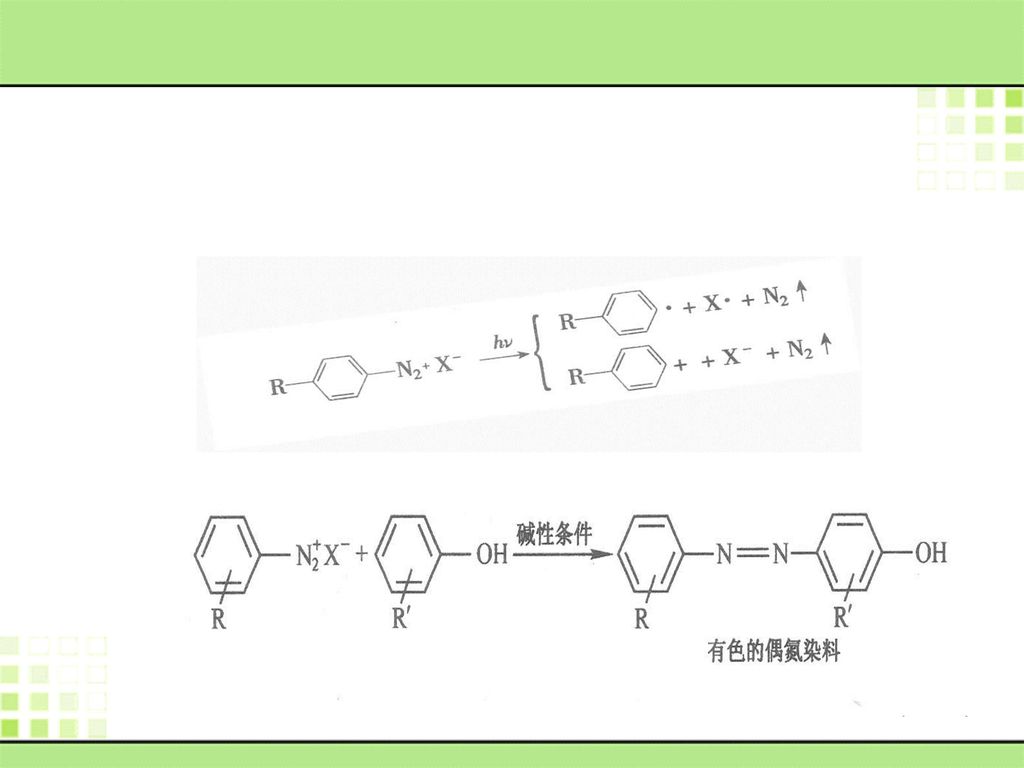
重氮盐的光敏性是其应用的基础。 重氮影像材料可分为染料影像材料(利用重氮盐在碱性条件下和酚类化合物因发生偶联反应而生成的偶氮燃料色度来显示影像的一类材料)、微泡影像材料(利用重氮盐光解后产生的氮气来成像的。
、微泡影像材料(利用重氮盐光解后产生的氮气来成像的。")
182
3.3自由基成像体系 自由基成像技术采用的是在光或离子辐照下使感光层中产生自由基,然后自由基与其它组分生成染料或破坏染料而成像的方法。
作为自由基成像体系的光活性物质多为有机卤化物,生成的自由基与感光层中其它组分发生反应,或生成染料(成色型)或破坏原有的染料(漂白型)。 成色型自由基成像体系大致可以分为三类,即缩合成像、氧化成像和聚合成像。
或破坏原有的染料(漂白型)。 成色型自由基成像体系大致可以分为三类,即缩合成像、氧化成像和聚合成像。")
183
缩合成像 CBr4 + hv ·CBr3 + ·Br

184
3.4 光子型信息存储的历史背景 多媒体技术的发展对计算机外存容量及速度不断提出更高的要求。
目前的计算机技术用复杂的硬件和软件涉及硬盘高速缓冲存储,但在随机存取大量数据时,在速度上并不能提供显著的提高。新一代的高密高速数字光存储技术的研究,为解决这一问题有很大的潜力。光信息存储已成为当今公认的当前科学技术领域的重大课题之一。

185
存储容量和数据传输率的发展趋势

186
3.5光子存储原理 光子存储的基本原理是基于光子同物质之间的直接相互作用,导致记录介质产生能够识别的物理和化学等性质的变化,从而达到信息记录的目的。 根据记录前后双稳态的特性既可以进行实时存储,又可以实现永久记忆。 目前研究的有四种不同的记录机制,即光子烧孔、电子俘获、光折变存储、光致变色存储。

187
3.5.1 光子烧孔 光子烧孔可分为光化学烧孔和光物理烧孔。
光化学烧孔是指光反应性分子分散在固体介质中,低温下,在激光诱导下发生具有位置选择性的光化学反应,引起在非均匀的宽带吸收光谱带上选择性地产生一个均匀的光谱孔。这是一种高密度存储方式,可在原来的二维存储中增加一个频率维度,从而提高其光存储密度。

188
光化学烧孔示意图

189
3.5.2电子俘获 电子俘获光存储是基于在存储材料中光诱导电子转移反应。组成这种材料的为至少包括两个具有不同的氧化还原电位的结构单元、原子、离子、分子或团簇,其中一个为电子给体(D),另一个为电子受体或空穴(A或H+)。
,另一个为电子受体或空穴(A或H+)。")
190
电子俘获存储材料能级示意图

191
3.6 光子存储的记录方式和特点 光子存储是基于光子同物质或记录介质的直接相互作用,这就决定了它的最大优点在于超高记录速度(很容易达到纳秒和皮秒数量级)、超高密度和超高分辨率。理论上分辨率可达分子水平,但实际达到的分辨率将由记录体系,包括激光波长、驱动装置、信号加工转换等过程所决定。当前技术条件下,二维(平面)记录密度的极限仍然在108—109bits/cm2。 光子存储记录方式有以下几种:斑点式、矢量式、图像式、全息式。
、超高密度和超高分辨率。理论上分辨率可达分子水平,但实际达到的分辨率将由记录体系,包括激光波长、驱动装置、信号加工转换等过程所决定。当前技术条件下,二维(平面)记录密度的极限仍然在108—109bits/cm2。 光子存储记录方式有以下几种:斑点式、矢量式、图像式、全息式。")
192
3.7 可擦重写光致变色光盘的工作原理

193
俘精酸酐光盘洋盘结构

194
4.1光致变色的定义 光致变色现象(photochromism)是指一个化合物(A)在受到一定波长的光照射时。可进行特定的化学反应,获得产物(B),由于结构的改变导致其吸收光谱发生明显的变化。而在另一波长的光照射下或热的作用下,又能回复到原来的形式。
是指一个化合物(A)在受到一定波长的光照射时。可进行特定的化学反应,获得产物(B),由于结构的改变导致其吸收光谱发生明显的变化。而在另一波长的光照射下或热的作用下,又能回复到原来的形式。")
195
光致变色反应及其吸收光谱示意图

196
光致变色反应势能曲线示意图 绝大多数光致变色体系是建立在单分子反应基础上的,势能面曲线的变化能更直观地表现出这一过程。图中a为基态势能曲线或叫热异构化势能曲线,b为化合物A的激发态势能曲线,c为化合物B的激发态势能曲线。

197
4.2目前对光致变色认识的深入 随着科学研究的发展和深入,基于单分子反应体系的光致变色定义显然是不完全的,需要补充,即目前光致变色的研究还应该包括以下三种模式: 1、多组分反应模式。 2、环式反应模式或多稳态可逆反应模式。 3、多光子光致变色反应体系。

198
4.2.1多组分反应模式 两个(A或B)或两个以上(很少见)的反应组分在光的作用下产生一种或多种产物(P),这种反应也必须是可逆的。
或两个以上(很少见)的反应组分在光的作用下产生一种或多种产物(P),这种反应也必须是可逆的。")
199
4.2.2 环式反应模式或多稳态可逆反应模式

200
4.2.3 多光子光致变色反应体系

201
4.2.4 光致变色分类总结

202
4.3 光致变色研究的发展历史 1867年Fritsche观察到黄色的并四苯在空气和光作用下的褪色现象,所生成的物质受热时重新生成并四苯。
1899年Markwald称之为光致变色(phototropy)。 从1940年起,人们为了弄清楚光致变色的机理、产物结构、中间体的形成、疲劳的产生而开展了大量的工作。这期间的工作主要集中在二苯乙烯、偶氮化合物等的顺反异构化的研究工作中。 20世纪50年代Hirshberg提出把上述现象称为“photochromism”,即光致变色现象。 1955年以后,军事及商业兴趣促使人们对光致变色进行研究。同时新型实验手段的出现也提高了人们对光致变色想象的认识。 目前对光致变色的研究大都集中在俘精酸酐、二芳基乙烯、螺吡喃等上,并继续探索和发现具有实际应用价值的研究体系。 热稳定性和耐疲劳性影响光致变色材料应用的两个主要因素。
。 从1940年起,人们为了弄清楚光致变色的机理、产物结构、中间体的形成、疲劳的产生而开展了大量的工作。这期间的工作主要集中在二苯乙烯、偶氮化合物等的顺反异构化的研究工作中。 20世纪50年代Hirshberg提出把上述现象称为 photochromism ,即光致变色现象。 1955年以后,军事及商业兴趣促使人们对光致变色进行研究。同时新型实验手段的出现也提高了人们对光致变色想象的认识。 目前对光致变色的研究大都集中在俘精酸酐、二芳基乙烯、螺吡喃等上,并继续探索和发现具有实际应用价值的研究体系。 热稳定性和耐疲劳性影响光致变色材料应用的两个主要因素。")
203
4.4 主要有机光致变色体系 有机光致变色体系可以根据物质的反应类型分为以下几类:键的异构、键的均裂、质子转移互变异构、顺反异构、氧化还原反应、周环反应体系、光致变色化合物的酸致变色。

204
4.5 有机光致变色的应用 1、光信息存储 2、生物分子活性的光调控 3、光致变色超分子 4、露光计 5、自显影感光胶片和全息摄影材料
6、光计算 7、防护与装饰材料 8、印刷板和印刷电路 9、伪装材料 10、防伪和鉴伪

205
4.6 无机变色材料 无机变色材料主要集中在过渡金属氧化物体系中,主要有MoO3、WO3、V2O5、NiO、Rh2O3、Nb2O5

206
4.7无机变色应用举例

207
5.1 新型高分子光折变材料 光折变效应(photorefractive effect)是指光照引起材料折光指数改变的效应。
从化学组成上来看,目前已发现具有光折变效应的材料大体上可分为三类:无机晶体、半导体、有机/高分子光折变材料。 高分子材料非常易于掺杂多种功能组分,化学方法将功能组分键接到高分子骨架上也较容易实现。此外高分子材料加工性能优良,可方便制备成薄膜、体块等所需形状。因此,高分子光折变材料从1991年被报道至今,已经发展为一个大家族,并构成光折变材料中最具发展前景的一类材料

208
5.2 实现光折变效应的条件 实现光折变效应的四个环节是:光生载流子的产生、光生载流子的输运、空间电荷场的建立、折光指数的电光调制。
一个高分子材料要获得光折变效应,必须具备两个重要性质:光导性和电光效应,这要求四种组分不可缺少:光敏剂、电荷(空穴或电子)传输成分、陷阱中心、电光分子或非线性光学分子。 光折变效应的实现是材料中多种功能组分密切配合、协同作用的结果,这要求各功能组分间不仅要有很好的性能匹配,还要求有很好的相容性。
传输成分、陷阱中心、电光分子或非线性光学分子。 光折变效应的实现是材料中多种功能组分密切配合、协同作用的结果,这要求各功能组分间不仅要有很好的性能匹配,还要求有很好的相容性。")
209
光折变效应微观机制示意图

210
6.超分子光化学和光化学分子器件

211
7.半导体和纳米材料在光化学中的应用 光致变色、电致变色、光化学反应

212
光化学治理环境污染 光化学治污法主要是利用半导体的特殊电子结构和光化学性质对污染物进行降解、氧化或还原等,最终得到无害或可再利用的物质。
作为光化学治理污染的载体---半导体,它具有特殊的电子结构,在光激励下,产生电子空穴对分离,光生空穴是强氧化剂(1.0—3.5V),能氧化绝大多数物质,光生电子是强还原剂( V),能还原绝大多数物质。 在环境治理中得到应用的半导体有TiO2、ZnO和CdS等。
,能氧化绝大多数物质,光生电子是强还原剂( V),能还原绝大多数物质。 在环境治理中得到应用的半导体有TiO2、ZnO和CdS等。")
213
TiO2光催化性能在环境治理中的应用示意图

Similar presentations














》 2.1 比零小的数 龙都初级中学 彭生翔>")








