Download presentation
Presentation is loading. Please wait.

1
人體試驗委員會簡介 花蓮慈濟 IRB 蘇雅慧 組長 97年9月30日

2
人體試驗定義 醫療法第8條:所稱人體試驗,係指醫療機構依醫學理論於人體施行新醫療技術、藥品或醫療器材之試驗研究。
赫爾辛基宣言:所謂人體試驗之對象即包含任何可辨識之人體組織或資料。

3
Human Subject A living individual about whom an investigator (whether professional or student) conducting research obtains Data through intervention or interaction with the individual, or Identifiable private information -- HHS regulation at 45 CFR (d)
")
4
18 HIPAA Identifiers Names
Geographic subdivisions smaller than a State Dates (except year) directly related to patients Telephone numbers Fax numbers addresses Social security numbers (ID) Medical record numbers Health plan beneficiary number 10. Account numbers
 directly related to patients. Telephone numbers. Fax numbers. addresses. Social security numbers (ID) Medical record numbers. Health plan beneficiary number. 10. Account numbers.")
5
18 HIPAA Identifiers Certificate/License numbers
Vehicle identifiers and serial numbers Device identifiers and serial numbers Web URLs Internet Protocol (IP) address numbers Biometric identifiers, including finger and voice prints Full face photographic images and any comparable images Any other unique identifying number, characteristic, or code, except as permitted under HIPAA to re-identify data
 address numbers. Biometric identifiers, including finger and voice prints. Full face photographic images and any comparable images. Any other unique identifying number, characteristic, or code, except as permitted under HIPAA to re-identify data.")
6
人體研究倫理政策指引 中華民國96年7月17日 衛署醫字第 號公告 人體研究除法令規定外,凡以研究為目的,取得、分析、調查人體之組織或個人之行為、理念、生理、心理、社會、遺傳,以及醫學有關資訊之過程均屬之。 人體研究應本最佳之科學實證與假設規劃,在資料取得、分析處理與成果運用之過程中,非經受研究者同意,均不得揭露其個人隱私資料;並應盡最大之可能管控風險發生;對於研究過程中可能導致之損害,應有包括損害補救措施在內之妥善因應計畫。

7
人體試驗管理機制 研究 常規醫療 非人體試驗 人體試驗 基因重組實驗守則(國科會) 醫療法 動物保護法(農委會) 臨床前試驗 GLP
人類胚胎及胚胎幹細胞研究倫理政策指引 臍帶血收集及處理作業規範 非人體試驗 人體試驗 相關前置規範 研究用人體檢體採集與使用注意事項 基因重組實驗守則(國科會) 醫療法 人體研究倫理政策指引 動物保護法(農委會) 新醫療技術、新藥品、新醫療器材 人工生殖法 臨床前試驗 GLP 人類胚胎及胚胎幹細胞研究條例(草案) 計畫書 IRB 核准 試驗期間監督 試驗完成報告 基因產品GMP、GTP 命令終止 組織工程產品GTP+GMP 受試者保護、療效、安全性 新藥品臨床試驗 GCP+cGMP 新醫療器材臨床試驗 人體器官保存庫管理辦法(草案) 新醫療技術人體試驗 優良組織操作規範 GTP 基因治療 GTP 體細胞治療 GTP 人體試驗管理機制
 醫療法. 人體研究倫理政策指引. 動物保護法(農委會) 新醫療技術、新藥品、新醫療器材. 人工生殖法. 臨床前試驗 GLP. 人類胚胎及胚胎幹細胞研究條例(草案) 計畫書. IRB. 核准. 試驗期間監督. 試驗完成報告. 基因產品GMP、GTP. 命令終止. 組織工程產品GTP+GMP. 受試者保護、療效、安全性. 新藥品臨床試驗 GCP+cGMP. 新醫療器材臨床試驗. 人體器官保存庫管理辦法(草案) 新醫療技術人體試驗. 優良組織操作規範 GTP. 基因治療 GTP. 體細胞治療 GTP. 人體試驗管理機制.")
8
人體試驗委員會 為確保受試者的權益與福祉,依據國家及國際法規,人體試驗委員會有權同意、要求變更與不同意人體相關研究之進行。 (王,2008)
")
9
醫療機構人體試驗委員會組織及作業基準 行政院衛生署92.11.12 衛署醫字第○九二○二○二五○七號公告
提升醫療機構人體試驗委員會之功能。 建立獨立之審查機制。 確保受試者之權益。 增進人體試驗計畫審查之效率。

10
醫療機構人體試驗委員會組織及作業基準 第一章 總則 第二章 人體試驗委員會之組成及召開 第三章 人體試驗計畫之申請與審查 第四章 決定之形成
第一章 總則 第二章 人體試驗委員會之組成及召開 第三章 人體試驗計畫之申請與審查 第四章 決定之形成 第五章 監督及管理 第六章 紀錄 第七章 附則

11
The main tasks of the IRB/EC
WHO guideline To assess the scientific merits of the research To verify that the research is in conformity with the laws and regulation, especially in relation with the protection of human subjects To evaluate the ethical acceptability of the research

12
受試者保護機制

13
Basic Ethical Principles
Respect for persons: informed consent Beneficence: risk/benefit assessment Justice: subject selection

14
優良臨床試驗準則(GCP) 臨床試驗設計、執行、監測、稽核、記錄、分析、報告之標準,可確保數據與所報告的結果均為可信與正確,受試者的權利、完整性、與身份機密均被保護。 保障受試驗者之人權(倫理) 確認臨床試驗的可信度(科學)
")
15
優良臨床試驗準則(GCP) 效益最大化 風險最小化 (科學性) (倫理、安全性) 確保受試者的權利、安全與福祉 確保臨床試驗數據的可信度
人體試驗委員會應確保受試者之權利、安全,以及福祉受到保護,且對於易受傷害受試者之臨床試驗,應特別留意。

16
GCP適用對象 應遵守者 供查驗登記用之藥品臨床試驗 建議參考者 供學術研究用之藥品臨床試驗 其他有關人類安全與福祉之臨床研究

17
臨床試驗受試者招募原則DOH96.6.6 依藥品優良臨床試驗準則第八十三條訂定之。 臨床試驗受試者招募廣告,不得於國中以下校園內刊登。
招募廣告應經人體試驗委員會核准始得刊登。 招募廣告得刊載下列內容: 試驗主持人姓名及地址。 試驗機構名稱及地址。 試驗目的或試驗概況。 主要納入及排除條件。 試驗之預期效益。 受試者應配合事項。 試驗聯絡人及聯絡方式。

18
臨床試驗受試者招募原則DOH96.6.6 招募廣告不得有下列內容或類似涵意之文字: 宣稱或暗示試驗藥品為安全、有效或可治癒疾病。
宣稱或暗示試驗藥品優於或相似於現行之藥物或治療。 宣稱或暗示受試者將接受新治療或新藥品,而未提及該研究屬試驗性質。 強調受試者將可獲得免費醫療或費用補助。 強調臨床試驗已經衛生主管機關或人體試驗委員會核准。 使用名額有限、即將截止或立即聯繫以免向偶等文字。 使用含有強制、引誘或鼓勵性質之圖表、圖片或符號。 其他經中央衛生主管機關公告不得刊登之內容。

19
試驗主持人資格 應藉由教育、訓練課程、和具備適當執行人體試驗的經驗之合格臨床醫師。倘若試驗主持人為非合格之臨床醫師,則需有一位共(協)同主持人為合格之臨床醫師,其應負責所有臨床試驗相關之醫療決定。 (倘若研究方式為研究個人或群體的特質或行為,或研究涉及調查,訪談,口述歷史,特定族群,計畫評估,人為因素評估或品質保證方法等,則試驗計畫主持人須為具獨立研究能力者即可) 所有人體試驗計畫主持人及相關研究團隊成員,皆需接受人體試驗相關教育訓練講習。 (本院ISO規定)
同主持人為合格之臨床醫師,其應負責所有臨床試驗相關之醫療決定。 (倘若研究方式為研究個人或群體的特質或行為,或研究涉及調查,訪談,口述歷史,特定族群,計畫評估,人為因素評估或品質保證方法等,則試驗計畫主持人須為具獨立研究能力者即可) 所有人體試驗計畫主持人及相關研究團隊成員,皆需接受人體試驗相關教育訓練講習。 (本院ISO規定)")
20
初審案送審管理 範圍:提送本委員會初次審查之計畫案,包括一般審查、簡易審查與追認審查。 依送審文件清單內容依序排列裝制成冊,首頁編目錄與頁碼
一般審查與簡易審查案件須檢附之文件資料為一式四份(一份正本三份影本),已通過其他人體試驗委員會核准且有同意證明的追認審查,則備妥原資料送審
,已通過其他人體試驗委員會核准且有同意證明的追認審查,則備妥原資料送審.")
21
計畫送審文件 人體試驗計畫初審案審查申請書 (內含最新履歷) 計劃主持人(協同主持人)聲明書 計畫書摘要
計畫書含參考文獻(註明版本與日期並簽名) 受試者同意書(註明版本與日期並簽名) 臨床試驗受試者同意書 研究用檢體採集與病歷資料提供同意書 受訪(問卷)同意書
 受試者同意書(註明版本與日期並簽名) 臨床試驗受試者同意書. 研究用檢體採集與病歷資料提供同意書. 受訪(問卷)同意書.")
22
計畫送審文件 研究問卷調查表或調查工具(視計畫需求檢附) 招募受試者資料,如廣告文宣或說明(視計畫需求檢附)
個案報告表、主持人手冊(視計畫需求檢附、新藥/醫療器材試驗適用) 受試者保險投保書、廠商許可執照、藥品/醫療器材許可證與相關仿單或說明(試驗藥品、醫療技術/器材須檢附) 臨床試驗協議書(廠商委託案件須檢附) 計畫主持人/共/協同主持人/研究成員參加臨床試驗相關訓練課程證書影本(須為最近3年內之證明)
 受試者保險投保書、廠商許可執照、藥品/醫療器材許可證與相關仿單或說明(試驗藥品、醫療技術/器材須檢附) 臨床試驗協議書(廠商委託案件須檢附) 計畫主持人/共/協同主持人/研究成員參加臨床試驗相關訓練課程證書影本(須為最近3年內之證明)")
23
計畫送審文件 其他相關資料 相同計畫經中央衛生主管機構或其他查委員審查之結果,不核准或須修正者,應檢附不核准理由或須修正內容
符合免受試者同意書之計畫案,須檢附免除「受試者同意書」申請書 符合簡易審查之案件,須檢附「簡易審查案件範圍勾選表」

24
簡易審查範圍 從手指、腳跟、耳朵採血或靜脈穿刺收集血液檢體 為研究目的,以前瞻性的非侵入性方法收集生物檢體
排除放射線或微波的使用,經由臨床上非侵入性方式(不涉及全身麻醉或鎮靜劑)所蒐集的資料。所使用的醫療器材(含適應症)須經中央衛生主管機關核准上市。 研究為例行臨床治療或診斷所收集之資料、文件、記錄、病理標本。 為研究目的蒐集的錄音、錄影、數位或影像資料記錄。 研究個人或群體的特質或行為(例如感覺、認知、動機、認同、語言、溝通、文化信仰或習慣和社會行為等),或研究涉及調查,訪談,口述歷史,特定族群,計畫評估,人為因素評估或品質保證方法等。
所蒐集的資料。所使用的醫療器材(含適應症)須經中央衛生主管機關核准上市。 研究為例行臨床治療或診斷所收集之資料、文件、記錄、病理標本。 為研究目的蒐集的錄音、錄影、數位或影像資料記錄。 研究個人或群體的特質或行為(例如感覺、認知、動機、認同、語言、溝通、文化信仰或習慣和社會行為等),或研究涉及調查,訪談,口述歷史,特定族群,計畫評估,人為因素評估或品質保證方法等。")
25
Exceptions for expedited review
Recommendations from FERCAP Sensitive (privacy and confidentiality) issues Vulnerable subjects
 issues. Vulnerable subjects.")
26
Vulnerable subjects Those groups that may contain some individuals who have limited autonomy: institutionalized, low socioeconomic, elderly, terminally ill, student, employees groups. Children, mentally incapacitated, individuals with dementia and other cognitive disorders, prisoners. Pregnant women.

27
審查流程 主持人備妥文件送審,由承辦人員先作行政審查,確認文件齊備才收案
一般審查:一位醫療專業委員、一位非醫療專業委員與一位院外審查專家進行初審,審查結果獲推薦,再送委員會會議裁決,通過後才核發同意函。 簡易審查:一位醫療專業委員與一位非醫療專業委員進行初審 ,審查結果獲推薦,即可先核發執行同意函 ,再送委員會會議核備。 追認案:一位醫療專業委員審查,審查通過之追認案行文通知不再核發同意函

28
計畫審查準則 試驗計劃設計與執行 計劃主持人的資格及經驗之適當性。 試驗執行機構之適當性,包括相關人員、設施與處理緊急狀況的能力。
試驗設計合乎科學要求且周詳。 選擇對照組之合理性。 受試者提前退出試驗的條件與退出後的照護是否適當。 監測與稽核試驗進行之規定是否充足,是否組成資料安全監測委員會。 預期風險與預期效益相較之合理性。 試驗結果之報告與發表方式對受試者之身份是否保密。 暫停或終止試驗的條件是否適當。

29
計畫審查準則 潛在受試者之招募 潛在受試者之母群體族群特性(包括性別、年齡、教育程度、文化背景、經濟狀況、種族淵源)。
受試者招募方式之適當性,廣告品、補助費等應符合公平、誠實、合適等原則。 將試驗資訊傳達給受試者的方式。 受試者納入條件之適當性。 受試者排除條件之適當性。 為試驗目的而取消或暫停標準治療之合理性。

30
計畫審查準則 受試者照護 試驗期間及試驗後,提供受試者的醫療照護是否適當(計畫結束後,是否有提供受試者繼續取得試驗產品之計畫)。
對受試者的心理支持與精神上的照護是否足夠。 受試者於試驗執行中自願提前退出時,所採取的處理方式是否合理。 試驗產品延長使用、緊急使用及恩慈(同情)使用標準是否合理。 受試者參與試驗之報酬是否合理。 受試者因參與試驗而受傷、殘障或死亡時之補償與治療應合理並於受試者同意書中載明。
使用標準是否合理。 受試者參與試驗之報酬是否合理。 受試者因參與試驗而受傷、殘障或死亡時之補償與治療應合理並於受試者同意書中載明。")
31
計畫審查準則 受試者同意取得程序 是否符合免簽署受試者同意書。 是否有取得受試者同意書的完整程序及負責人員。
告知受試者或法定代理人的資訊是否完整、充足與適當(同意書與試驗說明書內容是否清楚易懂)。 對無法親自行使同意權的受試者,取得合法代理人同意的理由與方式是否適當。 是否確保受試者於試驗執行期間能即時獲得與其權利、安全與福祉相關的最新資訊。 是否有受試者或其代理人投訴的管道與回應機制。
。 對無法親自行使同意權的受試者,取得合法代理人同意的理由與方式是否適當。 是否確保受試者於試驗執行期間能即時獲得與其權利、安全與福祉相關的最新資訊。 是否有受試者或其代理人投訴的管道與回應機制。")
32
計畫審查準則 隱私保護 是否有記載可能接觸受試者資料(含醫療紀錄與檢體)之人。 是否有提供為確保受試者隱私與個人資訊安全之措施。
若為社區試驗,是否有說明試驗對社區的影響與貢獻,及與社區協商的過程。

33
審查流程 一般審查 簡易審查 委員會會議審議裁決 (二個月一次) 核發同意函 獲推薦 通過 初審委員審查(10天) 主持人修正或申覆
(二週) 初審委員複審 未獲推薦 簡易審查 初審委員審查(10天) 主持人修正或申覆 (二週) 初審委員複審 未獲推薦 獲推薦 核發同意函 通過 委員會會議核備
 初審委員複審. 未獲推薦. 簡易審查. 初審委員審查(10天) 主持人修正或申覆. (二週) 初審委員複審. 未獲推薦. 獲推薦. 核發同意函. 通過. 委員會會議核備.")
34
變更案審查 適用於本委員會已核准之計畫案,任何內容之變更,計畫主持人必須向本委員會申請變更。變更案需經委員會審查通過才能再度開始執行。
變更案申請文件為一式四份,內容修改處必須以「粗體+底線」標示。 計畫變更申請書 計畫修改前後對照表 修改前、後相關文件,包括計畫書、受試者同意書、或個案報告表等

35
期中報告 呈送衛生署核備之計劃案每三個月須繳交執行情形報告送衛生署核備,其他計劃案每半年繳交。 計畫主持人應於接獲通知二週內繳交報告
期中報告表 收錄個案名單 受試者同意書簽名頁影本 若曾發生任何不良事件,須另附嚴重不良事件通報紀錄

36
嚴重不良事件(SAE)通報 死亡 危及生命 導致住院 造成永久性殘疾 延長住院 需作處置以防永久性害 先天性畸形 其他
嚴重不良事件(Serious Adverse Event),簡稱SAE SAE包括受試者發生: 死亡 危及生命 導致住院 造成永久性殘疾 延長住院 需作處置以防永久性害 先天性畸形 其他
,簡稱SAE. SAE包括受試者發生: 死亡. 危及生命. 導致住院. 造成永久性殘疾. 延長住院. 需作處置以防永久性害. 先天性畸形. 其他.")
37
嚴重不良事件(SAE)通報 計畫主持人或試驗委託者必須於事件發生15天內依衛生署規定向主管機關及本委員會完成通報,若為死亡及危及生命事件,則須於7天內通報 若為非預期的嚴重不良事件,計畫主持人須立即通報本委員會與主管機關。

38
結案報告 計畫主持人應於計畫案結束2個月內繳交結案報告 結案報告表 結案報告書 收錄個案名單 受試者同意書簽名頁影本
若曾發生任何不良事件,須另附嚴重不良事件通報紀錄

39
IRB 網站

41
花蓮慈院 IRB網站

43
CITI受試者保護訓練課程

44
The Collaborative Institutional Training Initiative (CITI Program)
CITI Program是關於受試者保護的網路訓練課程,經聯合人體試驗委員會(JIRB)的努力引進國內。不僅可做為人體試驗委員會之委員訓練,也可提供研究 醫師或研究護士等研究相關人員訓練之用。CITI Program內容由許多module 組成。每個module長短不一,約10-20分鐘可完成一個Module。最後有小考(quiz),通過小考該module才算完成。未通過的module可不限次數重覆學習。 每次小考(quiz)內容不全相同。
的努力引進國內。不僅可做為人體試驗委員會之委員訓練,也可提供研究 醫師或研究護士等研究相關人員訓練之用。CITI Program內容由許多module 組成。每個module長短不一,約10-20分鐘可完成一個Module。最後有小考(quiz),通過小考該module才算完成。未通過的module可不限次數重覆學習。 每次小考(quiz)內容不全相同。")
45
步驟1. Register 步驟1.

46
步驟2. Institution選Participating Institutions內的Joint Institutional Review Board-Taiwan

47
步驟3. 鍵入自定之Username和password

48
步驟4. 填入姓名和

49
步驟5. 填入個人基本資料。 Department請務必填上醫院名稱以利聯絡

50
步驟6. 選擇適合您狀況的module群組

51
步驟7. 選擇Grade book依序完成module。未通過小考(quiz)的module可不限次數重覆學習。小考(quiz)每次內容不全相同。
的module可不限次數重覆學習。小考(quiz)每次內容不全相同。")
52
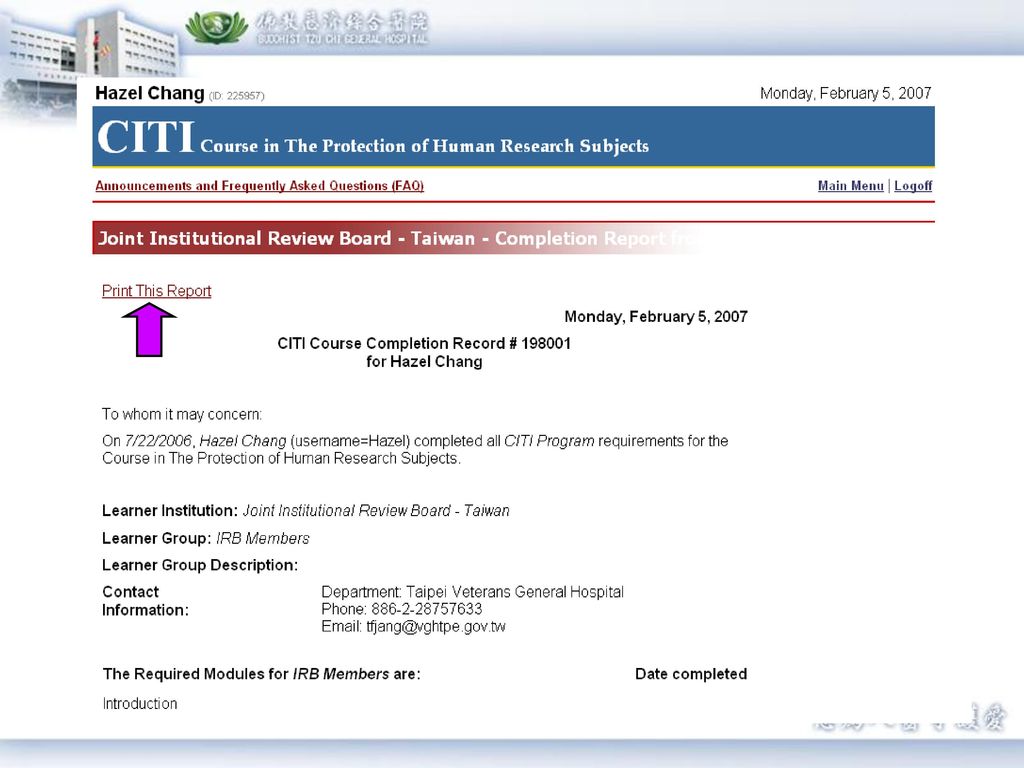
步驟8. 完成後請選擇complete reports form,並點選證書號碼自行印出證書。證書上會有基本資料和完成之Module清單。

54
感 恩 Thanks 一起為保護受試者努力!

Similar presentations







 □ 2.屋脊裝飾物審議 □ 3.裝飾柱/裝飾版審議>")




條例》的釋義、原則及主要條文>")
>")



>")


 DomainIII教育訓練>")