Download presentation
1
丁达尔效应 00-8-1

2
前言 1.胶体化学发展简史: A.1861年,Thomas Graham提出的胶体
起因:糖、无机盐等能结晶;胶体类Al(OH)3,明胶等不能结晶. B.40年后,韦曼的发现. 2.胶体化学所研究的对象: A.基本特性;B.由基本特性所决定的光学,电学,动力学性质; C.基本特性产生的原因,影响因素,变化规律; 00-8-1
3,明胶等不能结晶. B.40年后,韦曼的发现. 2.胶体化学所研究的对象: A.基本特性;B.由基本特性所决定的光学,电学,动力学性质; C.基本特性产生的原因,影响因素,变化规律;")
3
前言 3.胶体化学同医药学的关系: A.杀菌:如果用蛋白银溶胶作杀菌剂,不但有强烈的杀菌功效,还能避免对肌体的刺激作用;
B.防腐:将硝酸汞、明胶和氯化钠的混合物制成胶态甘汞,就比甘汞粒粉具有较高的防腐作用; C.药物的稳定性:除易引起变质的有害胶体,注意配伍禁忌即溶胶的相互凝结。另外制药工艺中的吸附、脱色、离子交换、层析技术等。 00-8-1

4
第一节.溶胶的分类及特征 一.溶胶的分类: 1.分散体系(disperse system):颗粒大小不等,形状不同的物体分散在均匀介质中所形成的体系. 分散质(dispersed phase):被分散的物质,粒子间有间隔,或称不连续相,相当于溶液中的溶质; 分散介质(dispersion medium):有分散相在其中的均匀介质,或称连续相,相当于溶液中的溶剂. 00-8-1
:有分散相在其中的均匀介质,或称连续相,相当于溶液中的溶剂")
5
第一节.溶胶的分类及特征 引言 2.分散系统的分类(按粒子大小)
溶液(或真溶液): 粒子直径小于1nm, 均相系统, 透明, 不能发生光的散射, 扩散速度快, 为热力学稳定状态. 胶体分散系统: 分散质在某方向上的直径在1~1000nm之间,可透明或不透明, 均可发生光散射, 胶体粒子扩散速率慢, 不能透过半透膜, 有较高的渗透压. 粗分散系统: 分散相粒子在某方向上的直径大于1000nm,如悬浮液, 乳状液, 泡沫, 粉尘等. 与胶体有许多共同特性. 00-8-1
: 粒子直径小于1nm, 均相系统, 透明, 不能发生光的散射, 扩散速度快, 为热力学稳定状态. 胶体分散系统: 分散质在某方向上的直径在1~1000nm之间,可透明或不透明, 均可发生光散射, 胶体粒子扩散速率慢, 不能透过半透膜, 有较高的渗透压. 粗分散系统: 分散相粒子在某方向上的直径大于1000nm,如悬浮液, 乳状液, 泡沫, 粉尘等. 与胶体有许多共同特性")
6
第一节.溶胶的分类及特征 本章主要介绍以液体为分散介质的系统, 重点是液溶胶(简称溶胶), 其次是乳状液, 泡沫等.
3..分散系统按聚集状态分类 分散介质 分散相 名称 实例 气 液 固 气溶胶 云, 雾, 喷雾 烟, 粉末 泡沫 乳状液 液溶胶或悬浮液 肥皂泡沫 牛奶, 含水原油 金溶胶, 油墨, 泥浆 固溶胶 泡沫塑料 珍珠, 蛋白石 有色玻璃, 某些合金 本章主要介绍以液体为分散介质的系统, 重点是液溶胶(简称溶胶), 其次是乳状液, 泡沫等. 00-8-1
, 其次是乳状液, 泡沫等")
7
第一节.溶胶的分类及特征 4.胶体系统又可分为三类:
溶胶: 高度分散的多相系统, 分散相与分散介质之间亲和力较弱, 有很大的相界面, 很高的表面能, 能自动聚集成大颗粒, 自动吸附某种离子而带电, 是热力学不稳定系统. 液溶胶传统上称憎液溶胶. 高分子溶液: 由于高分子以分子形式溶于介质中, 分散相与分散介质之间没有相界面, 大多是均相的热力学稳定系统. 传统上称亲液溶胶. 缔合胶体: 分散相是由表面活性剂缔合形成的胶束. 通常以水为分散介质, 胶束中表面活性剂的亲油基团向里, 亲水基团向外, 分散相与分散介质之间有很好的亲和性, 也是一类均相的热力学稳定系统. 有时称胶体电解质. 00-8-1

8
第一节.溶胶的分类及特征 二.溶胶的特征: A.多相的聚集体:(Heterogeneous polymerization)
胶体粒子为上千或上万的小分子的聚集体; B.高分散度:(High dispersity) 粒子小,A大,a大,dG大 C.聚结不稳定性:(Accumulation unstability)分散相粒子有自动聚集趋势,溶胶的这种性质称为聚结不稳定性。 00-8-1
 粒子小,A大,a大,dG大. C.聚结不稳定性:(Accumulation unstability)分散相粒子有自动聚集趋势,溶胶的这种性质称为聚结不稳定性。")
9
第二节溶胶的制备与净化 一.溶胶的制备: 1.制备的条件和途径: A.条件: ①难溶; ②加入合适的稳定剂. B.途径:
①分散法(dispersion method ):进一步粉碎粗分散系中的大粒子,提高分散度,这种方法称为分散法。 ②凝聚法(condensation method):设法使一般溶液中的溶质分子聚集成多分子聚集体,降低分散度至粒子大小达到 m,这种方法称为凝聚法 C.均匀胶体的制备 00-8-1
:进一步粉碎粗分散系中的大粒子,提高分散度,这种方法称为分散法。 ②凝聚法(condensation method):设法使一般溶液中的溶质分子聚集成多分子聚集体,降低分散度至粒子大小达到 m,这种方法称为凝聚法. C.均匀胶体的制备")
10
①分散法 胶体系统 1~1000nm 分子分散系统 < 1nm 粗分散 系统 >1000nm
由小变大 凝聚法 由大变小 分散法 粗分散 系统 >1000nm 电弧法 胶体磨 喷射磨 物理凝聚法 化学凝聚法 蒸气凝聚法 过饱和法 分散法利用机械设备将粗分散物料分散成胶体. 分散过程所消耗的机械功或电功远大于系统表面吉布斯函数的增加, 大部分能量以热量耗散. 分散法常用下列设备: 胶体磨 主要部件是一高速转动的圆盘, 转速5000~10000 rpm, 圆盘与外壳间的微隙可调至约510-6m. 湿磨的粉碎程度更高. 粉碎时常加入少量表面活性剂以防止微粒聚集. 00-8-1

11
①分散法 气流粉碎机(又称喷射磨): 主要部件是在粉碎室的边缘上装有与周边成一定角度的两个高压喷嘴, 将两股高压气流及物料以大约音速的速率喷入粉碎室, 气流形成涡流, 粒子相互碰撞, 摩擦及剪切作用而被粉碎. 优点是效率高, 粉碎程度可达10-6m以下, 混入杂质少. 电弧法: 将欲分散的金属作为电极, 浸入水中, 通入直流电, 调节两极距离使产生电弧. 电弧温度很高, 而使电极表面的金属气化, 金属蒸气遇水冷却而冷凝成胶体系统. 00-8-1

12
物理凝聚法: 将蒸气或溶解状态的物质凝聚成胶体的方法.
②凝聚法 物理凝聚法: 将蒸气或溶解状态的物质凝聚成胶体的方法. 蒸气凝聚法 采用特制的仪器, 使金属和有机溶剂在超低压下蒸发, 而后冷凝而成金属的有机溶胶. 过饱和法 更换溶剂或者降温冷却, 使溶质溶解度降低至过饱和, 然后从溶液中结晶而聚集成溶胶. 如制备硫的水溶胶, 树脂或脂肪的水溶胶, 苯的水溶胶, 硫的醇溶胶等. 化学凝聚法: 通过控制化学反应中不溶性产物的析晶过程, 使其停留在胶核尺度阶段, 而得到溶胶的方法. 一般采用较大的过饱和度和较低的温度以利于晶核的大量形成而减缓晶体长大的速度, 防止难溶性物质的聚沉. 00-8-1

13
(a) 利用FeCl3的水解反应,生成Fe(OH)3溶胶 FeCl3 + 3H2O Fe(OH)3+3HCl
②凝聚法 例如: (a) 利用FeCl3的水解反应,生成Fe(OH)3溶胶 FeCl3 + 3H2O Fe(OH)3+3HCl 在不断搅拌下, 将FeCl3稀溶液滴入沸腾的水中, 即可生成棕红色, 透明的Fe(OH)3溶胶. 过量的FeCl3同时又起到稳定剂的作用, Fe(OH)3的微小晶体选择性地吸附Fe3+离子, 可形成带正电荷的胶体粒子. (b) 利用As2O3的复分解反应制备硫化砷溶胶: As2O3 + 3H2O 2H3AsO3 2H3AsO3 + 3H2S As2S3 + 6H2O 在As2O3的饱和或过饱和的水溶液中, 缓慢通入H2S气体, 即可生成淡黄色As2S3溶胶. HS-为稳定剂, 胶粒带负电荷. 00-8-1
 利用FeCl3的水解反应,生成Fe(OH)3溶胶. FeCl3 + 3H2O Fe(OH)3+3HCl. 在不断搅拌下, 将FeCl3稀溶液滴入沸腾的水中, 即可生成棕红色, 透明的Fe(OH)3溶胶. 过量的FeCl3同时又起到稳定剂的作用, Fe(OH)3的微小晶体选择性地吸附Fe3+离子, 可形成带正电荷的胶体粒子. (b) 利用As2O3的复分解反应制备硫化砷溶胶: As2O3 + 3H2O 2H3AsO3. 2H3AsO3 + 3H2S As2S3 + 6H2O. 在As2O3的饱和或过饱和的水溶液中, 缓慢通入H2S气体, 即可生成淡黄色As2S3溶胶. HS-为稳定剂, 胶粒带负电荷")
14
C.均匀胶体的制备 ①多级分散:含有大小不一的胶体粒子。 ②单级分散(monodispersion) :制备粒子大小一致的胶体粒子。
③形状:球形、棒性、立方形、椭圆形等各种均匀胶体。 电分散; ④应用:均匀胶体在特种陶瓷、催化剂、颜料、油墨、磁性材料及感光材料的研制和产品质量的提高等方面有着广泛的应用前景 00-8-1

15
二.溶胶的净化 溶胶的净化: 将溶胶制备过程中引入或产生的过量电解质或其它杂质除去, 以提高溶胶的稳定性和纯度.
渗析法: 净化溶胶的最常用方法. 利用胶粒不能透过半透膜的特点, 分离出溶胶中多余的电解质或其它杂质. 一般可用羊皮纸, 动物膀胱膜, 硝酸或醋酸纤维素等作为半透膜. 为加快渗透作用, 可加大渗透面积, 适当提高温度或加外电场. 电渗析: 利用外电场来大大加速正, 负离子定向渗透速度. 适宜的电解质是形成胶体系统必不可少的条件, 但过量的电解质的加入又是破坏胶体系统的有效方法. 因此, 要保持溶胶的稳定性, 必须除去多余的电解质. 00-8-1

16
第三节溶胶的动力学性质 一.布朗运动:用分子运动论的观点, 研究胶体粒子的无规则运动以及由此而产生的扩散, 渗透等现象, 研究胶粒在重力场作用下, 粒子浓度随高度的变化规律. 布朗(Brown)运动: 溶胶中的分散相粒子的不停息的和无规则的运动. 这种现象是植物学家(Brown)于1827年首先从水中悬浮花粉的运动中观察到的. 用超显微镜可以观察布朗运动. “布朗运动”动画 分散介质分子处于无规则的热运动状态, 从各个方向不断撞击分散相粒子. 布朗运动是分子热运动的必然结果, 是胶体粒子的热运动. 00-8-1
运动: 溶胶中的分散相粒子的不停息的和无规则的运动. 这种现象是植物学家(Brown)于1827年首先从水中悬浮花粉的运动中观察到的. 用超显微镜可以观察布朗运动. 布朗运动 动画. 分散介质分子处于无规则的热运动状态, 从各个方向不断撞击分散相粒子. 布朗运动是分子热运动的必然结果, 是胶体粒子的热运动")
17
一.布朗运动 1905年左右, 爱因斯坦用几率的概念和分子运动论的观点, 创立了布朗运动理论, 得出 爱因斯坦-布朗运动平均位移公式
— 在时间 t 内粒子沿 x 轴方向的平均位移; r — 粒子半径; η—介质粘度; L —阿佛加德罗常数. 斯威德伯格(Svedberg)用超显微镜对一定粒度的金溶胶进行摄影实验, 所得结果证实了上述公式的准确性, 有力地证明了分子运动论完全可以用于胶体分散系统. 用该公式测定分散相粒子的大小及阿伏加德罗常数, 得到了同样令人满意的结果. 00-8-1
用超显微镜对一定粒度的金溶胶进行摄影实验, 所得结果证实了上述公式的准确性, 有力地证明了分子运动论完全可以用于胶体分散系统. 用该公式测定分散相粒子的大小及阿伏加德罗常数, 得到了同样令人满意的结果")
18
二.扩散与渗透 一.)扩散: 在有浓度梯度时, 物质粒子因热运动而发生宏观上的定向迁移的现象. 产生的原因是物质粒子的热运动.
因分散相粒子的质量比一般分子大千百倍, 其扩散速率远小于一般分子的扩散速率(一般以扩散系数D来衡量). 胶体系统的扩散亦可用菲克第一定律描述: dn/dt = -DAs(dc /dx) 对于球形粒子的稀溶液, 且为单级分散(即粒子大小一定), 上式可用于测定扩散系数. 进而可求球形粒子的摩尔质量: 00-8-1
. 胶体系统的扩散亦可用菲克第一定律描述: dn/dt = -DAs(dc /dx) 对于球形粒子的稀溶液, 且为单级分散(即粒子大小一定), 上式可用于测定扩散系数. 进而可求球形粒子的摩尔质量:")
19
二)渗透 1.渗透压: 渗透平衡时, 溶剂液面和同一水平的溶液截面上所受的压力之差, 用 表示. 2.渗透发生的原因:
渗透平衡时, 溶液一方压力的变化对溶剂化学势的影响抵消了组成的影响, 从而有 3.范特霍夫渗透压公式 00-8-1

20
二)渗透 在定温下,溶液的渗透压与溶质的浓度成正比。溶液愈稀,公式愈准确 。 渗透压测定法常被用来测定生物体内大分子的摩尔质量。
渗透现象不仅在溶液与溶剂之间存在,在不同浓度的溶液中同样存在 等渗溶液:相等渗透压的溶液彼此称为等渗溶液; 高渗溶液:对于渗透压不相等的两种溶液,渗透压相对较高的叫高渗溶液; 低渗溶液:渗透压相对较低的叫做低渗溶液 。 等渗溶液在药学上有重要意义 如眼药水必须与眼球组织内的液体具有相同的渗透压,否则会引起疼痛; 静脉注射用的盐水与血液是等渗溶液 00-8-1

21
二)渗透 渗透原理被用来处理尿毒症. 在人工肾里, 病人的血液在玻璃软管(用作半透膜)循环, 血液里的小分子废物向管外渗透, 从而使血液得到净化. 纯水向红萝卜内渗透 渗析实验 00-8-1 例18

22
二)渗透 三)溶胶的渗透压(osmotic pressure)特征:
①一个分散相粒子(多分子聚集体)才是一个动力单位,所以一个溶胶粒子产生渗透压的作用只相当于一个普通分子; ②一定浓度的溶液,当溶质分子凝聚成胶体粒子后,粒子浓度要比原来分子浓度小几千至几百万倍,因此,溶胶的渗透压至多只是原来溶液的几千分之一; ③溶胶的聚结不稳定性,渗透压可随着聚结的进行而逐渐减小。 00-8-1
才是一个动力单位,所以一个溶胶粒子产生渗透压的作用只相当于一个普通分子; ②一定浓度的溶液,当溶质分子凝聚成胶体粒子后,粒子浓度要比原来分子浓度小几千至几百万倍,因此,溶胶的渗透压至多只是原来溶液的几千分之一; ③溶胶的聚结不稳定性,渗透压可随着聚结的进行而逐渐减小。")
23
三.沉降与沉降平衡 1.沉降:悬浮在液态介质中的密度比介质大的粒子受重力作用下沉的现象,叫做沉降。
F重力 r为粒子半径 粒子密度 0液态介质的密度 2.斯托克斯定律(Stokes law)为 f=6ru 为液态介质的粘度,u为粒子沉降速度。 3.等速沉降: 沉降开始后,粒子在重力作用下沉降加速进行,u越来越大,沉降阻力随之增大,当增大至沉降阻力等于粒子的重力时,粒子处于力的平衡状态w=f,此为等速沉降。 重力沉降速度 00-8-1
为. f=6ru. 为液态介质的粘度,u为粒子沉降速度。 3.等速沉降: 沉降开始后,粒子在重力作用下沉降加速进行,u越来越大,沉降阻力随之增大,当增大至沉降阻力等于粒子的重力时,粒子处于力的平衡状态w=f,此为等速沉降。 重力沉降速度")
24
三.沉降与沉降平衡 4.沉降平衡: 指分散相粒子的扩散速率与沉降速率相等的状态. 沉降与扩散是两个相反的互为竞争的过程,大质量的粒子易于沉降, 反之易于扩散. 在重力场中达到沉降平衡时, 粒子浓度随高度而变化的分布定律 C1, C2 分别为高度h1, h2 截面上粒子的浓度; , 0 分别为粒子及介质的密度; g 为重力加速度. NA:阿弗加德罗常数 上式不受粒子形状的限制, 但要求粒子大小相等. 一般分散系统为多级分散(粒子大小不等), 可用上式分别计算出不同大小粒子的分布. 粒子愈大, 其平衡浓度梯度愈大. 00-8-1
, 可用上式分别计算出不同大小粒子的分布. 粒子愈大, 其平衡浓度梯度愈大")
25
三.沉降与沉降平衡 由于溶胶分散相粒子的沉降与扩散速率皆很慢, 达到沉降平衡状态往往需要很长时间. 若系统温度发生变化, 所引起的对流作用也能阻碍平衡的建立. 上式也可用于计算大气压的高度分布. 将大气视为理想气体且不考虑气温的高度变化, 则不同高度处有 c2 / c1 = p2 / p1. 另大气粒子无须考虑浮力校正, 即 1-( 0 /) = 1, 所以有 式中M为空气的平均摩尔质量(29 10-3 kgmol-1 ). 上式也可用于各组分的计算, 如可算出298.15K 的大气层中氧气的分压降低一半的高度为 km. 显然愈靠近大地, CO2, SO2, NO2等分子量较大的气体的含量愈高. 00-8-1
 = 1, 所以有. 式中M为空气的平均摩尔质量(29 10-3 kgmol-1 ). 上式也可用于各组分的计算, 如可算出298.15K 的大气层中氧气的分压降低一半的高度为 km. 显然愈靠近大地, CO2, SO2, NO2等分子量较大的气体的含量愈高")
26
第四节溶胶的光学性质 一.丁铎尔效应:由于溶胶的高度分散性和多相不均匀性, 当一束波长大于溶胶分散相粒子尺寸的入射光照射到溶胶系统, 可发生散射现象-丁铎尔现象. 透镜 溶胶 丁铎尔效应 光源 00-8-1

27
一.丁铎尔效应 当入射光的频率与分子的固有频率相同时, 发生光的吸收. 当光束与系统不发生任何相互作用时, 则可透过.
当入射光的波长小于分散粒子的尺寸时, 则发生光的反射. 若入射光的波长大于分散粒子的尺寸时, 则发生光的散射. 可见光波长在400~700nm范围内, 大于一般胶体粒子的尺寸(1~100)nm, 可发生光的散射. 光的振动频率高达1015Hz, 光的照射相当于外加电磁场作用于胶体粒子, 使围绕分子或原子运动的电子被迫产生振动, 而质量达大于电子的原子核是无法跟上如此高频率的振动的, 这样被光照射的微小晶体上的每个分子, 便以一个次级光源的形式, 向四面八方辐射出与入射光有相同频率的次级光波. 丁铎尔现象的实质是光的散射作用. 丁铎尔效应又称为乳光效应, 散射光的强度可由瑞利公式计算. 00-8-1
nm, 可发生光的散射. 光的振动频率高达1015Hz, 光的照射相当于外加电磁场作用于胶体粒子, 使围绕分子或原子运动的电子被迫产生振动, 而质量达大于电子的原子核是无法跟上如此高频率的振动的, 这样被光照射的微小晶体上的每个分子, 便以一个次级光源的形式, 向四面八方辐射出与入射光有相同频率的次级光波. 丁铎尔现象的实质是光的散射作用. 丁铎尔效应又称为乳光效应, 散射光的强度可由瑞利公式计算")
28
二.雷利公式 假设: 粒子的尺寸远小于入射光波长; 粒子间距离较远, 可不考虑各个粒子散射光的相互干涉; 粒子不导电.
假设: 粒子的尺寸远小于入射光波长; 粒子间距离较远, 可不考虑各个粒子散射光的相互干涉; 粒子不导电. 雷利(L.W.Rayleigh)公式 I — 单位体积液溶胶的散射光强度; I0—入射光强度; l — 观察者与散射中心的距离; V — 分散相粒子的体积; ν — 单位体积中的粒子数; —入射光波长; — 散射角; n, n0 — 分别为分散相及分散介质的折射率; I V 2, 真溶液小分子体积微小, 丁铎尔效应微弱; 粗分散悬浮液粒子尺寸大于可见光波长, 无丁铎尔效应; 只有溶胶丁铎尔效应显著. 用丁铎尔效应可鉴别分散系统种类. 00-8-1
公式. I — 单位体积液溶胶的散射光强度; I0—入射光强度; l — 观察者与散射中心的距离; V — 分散相粒子的体积; ν — 单位体积中的粒子数; —入射光波长; — 散射角; n, n0 — 分别为分散相及分散介质的折射率; I V 2, 真溶液小分子体积微小, 丁铎尔效应微弱; 粗分散悬浮液粒子尺寸大于可见光波长, 无丁铎尔效应; 只有溶胶丁铎尔效应显著. 用丁铎尔效应可鉴别分散系统种类")
29
二.雷利公式 I -4, 波长愈短散射光愈强. 白光中蓝光和紫光散射最强, 红光最弱. 当用白光照射溶胶时, 在与入射光垂直方向呈淡蓝色, 而透过光呈现橙红色. 分散相与介质的折射指数相差越大, 散射光越强. 憎液溶胶分散相与介质之间有明显的界面, 其折射率相差较大, 乳光效应很强. 而高分子真溶液是均相系统, 乳光甚微, 故可依此来区别高分子溶液与溶胶. I C, 此关系可用来测量溶胶的浓度. 乳光强度又称为浊度, 浊度计就是根据这一原理设计的. 00-8-1

30
三.超显微镜与粒子大小的近似测定 在超显微镜下看到的是粒子的散射光的影像, 其大小比胶体粒子本身的投影大数倍之多. 粒子的平均大小可以估算.
如果已知单位体积溶胶中分散相的质量, 则由数密度可求出每个粒子的质量m, 假设粒子是半径为r 的球形, 粒子密度为, 则由 m = (4/3) r3 即可求得胶粒的平均半径: 超显微镜是根据丁铎尔效应而设计的可看到胶体粒子的存在及运动的显微镜. 与普通显微镜的差别是强光源照射, 在与入射光垂直的方向上及黑暗视野条件下观察. 超显微镜与电子显微镜:电子波长为0.005nm前者为虚影,(从侧面)后者为实影 5.溶胶的颜色: ①粒子的大小,越大,向长波长方向移动;②补色; ③以颜色可判断其溶胶的稳定性.. 00-8-1
 r3 即可求得胶粒的平均半径: 超显微镜是根据丁铎尔效应而设计的可看到胶体粒子的存在及运动的显微镜. 与普通显微镜的差别是强光源照射, 在与入射光垂直的方向上及黑暗视野条件下观察. 超显微镜与电子显微镜:电子波长为0.005nm前者为虚影,(从侧面)后者为实影. 5.溶胶的颜色: ①粒子的大小,越大,向长波长方向移动;②补色; ③以颜色可判断其溶胶的稳定性")
31
第五节 溶胶的电学性质 一.电动现象:(electrokinetic phenomena)
指溶胶粒子的运动与电性质之间的关系,包括:电泳、电渗、流动电势和沉降电势。 1. 电泳和电渗 1)电泳: (electrophonesis) :在外电场作用 下, 胶体粒子在分散介质中定向移动的现 象. 电泳现象表明胶体粒子是带电的. NaCl溶液 Fe(OH)3 溶胶 + - 电泳示意图 图中Fe(OH)3 溶胶在电场作用下向阴极方向移动, 证明Fe(OH)3的胶体粒子是带正电荷的. 对于半径r 较大, 双电层厚度较小的质点, 其表面可作为平面表面处理, 此时电泳速度 与 电势的关系为 00-8-1
电泳: (electrophonesis) :在外电场作用. 下, 胶体粒子在分散介质中定向移动的现. 象. 电泳现象表明胶体粒子是带电的. NaCl溶液. Fe(OH)3 溶胶. + - 电泳示意图. 图中Fe(OH)3 溶胶在电场作用下向阴极方向移动, 证明Fe(OH)3的胶体粒子是带正电荷的. 对于半径r 较大, 双电层厚度较小的质点, 其表面可作为平面表面处理, 此时电泳速度 与 电势的关系为")
32
一.溶胶的电动现象 2)电渗(electro-osmosis): : 在多孔膜(或毛细管)的两端施加一定电压, 液体(分散介质)将通过多孔膜而定向移动的现象(带电的固相不动). 导线管 多孔塞 + - 电极 毛细管 气泡 吹入一个气泡 水或溶液 电渗测定装置 如图所示, 通电后液体通过多孔塞而定向流动, 可从水平毛细管中小气泡的移动来观察循环流动的方向.若多孔塞阻力远大于毛细管阻力, 可通过小气泡在一定时间内移动的距离来计算电渗流的流速. 流动方向和流速大小与多孔塞材料(带电的固相), 流体性质以及外加电解质有关. 00-8-1
, 流体性质以及外加电解质有关")
33
一.溶胶的电动现象 3)流动电势: 在外力作用下, 迫使液体通过多孔隔膜(或毛细管)定向流动, 多孔隔膜两端所产生的电势差.它是电渗的逆现象. 4)沉降电势: 分散相粒子在重力场或离心力场的作用下迅速移动时, 在移动方向的两端所产生的电势差. 它是电泳的逆现象. N2加压 电位差计 电极 多孔塞 流动电势测量示意图 沉降电势测量示意图 00-8-1

34
二.粒子带电原因 1.胶核的选择吸附:溶胶粒子(胶核)是多分子聚集体,与介质之间有巨大界面,表面能很大,能选择吸附作为稳定剂的离子到界面上来。 里巴托夫规则(Liepatoff’s Rule):溶胶粒子选择吸附和它组成相同或相类似的离子。 2.固体表面上的物质粒子, 在溶液中发生电离.在某些胶体粒子的生成过程中,粒子表面上的一部分分子发生电离,把与粒子组成相类似的离子作为决定电位的离子吸附到表面上使粒子带电。 如:硅胶粒子为SiO2的多分子聚集体,表面上的SiO2 生成H2SiO3,H2SiO3是弱酸,部分电离出SiO32-离子; 如蛋白质中的氨基酸分子, 在pH值低时氨基形成-NH3+而带正电,在pH值高时羧基形成-COO-而带负电. 00-8-1

35
二.粒子带电原因 3.两相接触生电(摩擦带电):
溶胶的分散相粒子与分散介质若为介电常数不同的非导电物质,则互相接触时可因摩擦而生电。由于它们对电子的亲和力不同,电子可从亲和力较小的一相流入亲和力较大的一相,使粒子带电。科恩(Cohen)总结出一条经验规律:介电常数较大的一相带正电,另一相带负电。 4.晶格取代: 泥土粒子中,晶格中Al3+,Si4+被Ca2+,Mg2+取代后,整个粒子带正电,与原本电荷相比,正负电荷数不相同。 00-8-1
总结出一条经验规律:介电常数较大的一相带正电,另一相带负电。 4.晶格取代: 泥土粒子中,晶格中Al3+,Si4+被Ca2+,Mg2+取代后,整个粒子带正电,与原本电荷相比,正负电荷数不相同。")
36
三.扩散双电层理论 处在溶液中的带电固体表面, 主要由于静电吸引力的存在, 必然要吸引等电量的异电离子(或反离子)环绕在固体周围, 这样便在固液两相之间形成双电层. 1.双电层电容器模型(1879年亥姆霍兹提出): 正负离子整齐地排列于界面层的两侧, 电荷分布情况就如同平行板电容器, 两层间的距离很小, 与离子半径相当. 该模型过于简单, 与实际情况矛盾很多, 如不能解释带电质点的表面电势0 与质点运动时固液两相发生相对移动时所产生的电势差—— 电势(电动电势)的区别, 也不能解释电解质对 电势的影响等. x 亥姆霍兹双电层模型 00-8-1
: 正负离子整齐地排列于界面层的两侧, 电荷分布情况就如同平行板电容器, 两层间的距离很小, 与离子半径相当. 该模型过于简单, 与实际情况矛盾很多, 如不能解释带电质点的表面电势0 与质点运动时固液两相发生相对移动时所产生的电势差—— 电势(电动电势)的区别, 也不能解释电解质对 电势的影响等. x. 亥姆霍兹双电层模型")
37
三.扩散双电层理论 2.扩散双电层模型(1910年古依和查普曼提出): 溶液中的反离子因为热运动应呈扩散状态分布在溶液中, 而不是整齐地排列在一个平面上. 如图紧靠固体表面, 反离子浓度最大, 随着离开固体表面愈远, 反离子浓度降低, 形成一个反离子的扩散层.古依和查普曼假设, 在质点表面可看作无限大的平面, 表面电荷分布均匀, 溶剂介电常数到处相同的条件下, 距表面一定距离x 处的电势与表面电势0 的关系遵守: 古依的扩散双电层模型 0 x 玻耳兹曼定律: = 0 e -x 式中 的倒数具有双电层厚度的含义. 该模型没有考虑到反离子的吸附及离子的溶剂化, 未能反映界面紧密层的存在. 00-8-1

38
三.扩散双电层理论 3.斯特恩双电层模型(1924年Stern 提出): 该模型认为溶液一侧的带电层应分为紧密层和扩散层两部分.
紧密层: 溶液中反离子及溶剂分子受到足够大的静电力, 范德华力或特性吸附力, 而紧密吸附在固体表面上. 其余反离子则构成扩散层. 斯特恩面: 紧密层中反离子的电性中心所连成的假想面. 距固体表面的距离约为水化离子的半径.斯特恩面上的电势 称为斯特恩电势. 滑动面: 指固液两相发生相对移动的界面, 在斯特恩面稍外一些, 是凹凸不平的曲面.滑动面至溶液本体间的电势差称为 电势. 00-8-1

39
三.扩散双电层理论 电势 距离 滑动面 c1< c2 < c3 < c4 c1 c2 c3 c4 电解质浓度对 电势的影响 等电点: 当电解质浓度增大时, 介质中反离子的浓度加大而更多地进入滑动面内, 使扩散层变薄, 电势在数值上变小. 当电解质的浓度足够大时, 可使 电势为零. 此时的状态称为等电点. 处于等电点的粒子是不带电的, 电泳, 电渗的速度也必然为零, 溶胶非常易于聚沉. 电势只有在固液两相发生相对移动时才能呈现出来. 电势的大小反映了胶粒带电的程度, 其值越高表明胶粒带电越多, 扩散层越厚. 00-8-1

40
-电位的大小可衡量溶胶的稳定性。-电位的计算公式为
电泳的计算 -电位的大小可衡量溶胶的稳定性。-电位的计算公式为 u 为电泳的速度 E为电位梯度 εr是介质的介电常数 是介质的粘度 K是一个常数 其值与胶粒的形状有关:球形粒子K=6,棒状粒子K=4。 -电位的正付与决定电位的离子符号相同。 00-8-1

41
第六节溶胶的稳定性 一.溶胶的胶团结构 胶核: 由分子, 原子或离子形成的固态微粒及其吸附的离子所组成的部分.
胶体粒子: 滑动面所包围的带电体, 包括胶核及一部分被吸附的反离子. 胶团: 整个扩散层及其所包围的电中性体, 包括胶体粒子和扩散层中的那部分过剩反离子. 如在稀AgNO3溶液中缓慢加入少量KI稀溶液, 得到AgI溶胶(正溶胶), 过剩的AgNO3则起稳定剂的作用. 胶团结构式为: [(AgI)m nAg+(n-x)NO3-] x+ xNO3- 胶核 胶粒 滑动面 胶团 00-8-1
, 过剩的AgNO3则起稳定剂的作用. 胶团结构式为: [(AgI)m nAg+(n-x)NO3-] x+ xNO3- 胶核. 胶粒. 滑动面. 胶团")
42
一.溶胶的胶团结构 NO3- (AgI)m Ag + AgI 胶团示意图 00-8-1
m Ag + AgI 胶团示意图")
43
一.溶胶的胶团结构 如在稀KI溶液中缓慢加入少量AgNO3稀溶液, 得到AgI溶胶(负溶胶), 过剩的KI则起稳定剂的作用. 胶团结构式为:
[ (AgI)m nI- (n-x)K+] x- xK+ 胶核 胶粒 胶团 如在稀KI溶液中缓慢加入少量AgNO3稀溶液, 得到AgI溶胶(负溶胶), 过剩的KI则起稳定剂的作用. 胶团结构式为: 再如SiO2溶胶, SiO2微粒与水生成弱酸H2SiO3, 电离出的 SiO32-有一部分吸附在SiO2微粒表面上, 形成带负电的胶核, H+为反离子. 反应过程可表示为 胶团结构可表示为 [(SiO2)m nSiO32-2(n-x) H +] 2x-2xH+ 在同一个溶胶中, 每个固体微粒所含分子个数 m及其所吸附的离子个数 n 都不一定是相等的. 但整个胶团是电中性的, 总电荷数应为零, 书写胶团结构时要注意这一点. 00-8-1
m nI- (n-x)K+] x- xK+ 胶核. 胶粒. 胶团. 如在稀KI溶液中缓慢加入少量AgNO3稀溶液, 得到AgI溶胶(负溶胶), 过剩的KI则起稳定剂的作用. 胶团结构式为: 再如SiO2溶胶, SiO2微粒与水生成弱酸H2SiO3, 电离出的 SiO32-有一部分吸附在SiO2微粒表面上, 形成带负电的胶核, H+为反离子. 反应过程可表示为. 胶团结构可表示为 [(SiO2)m nSiO32-2(n-x) H +] 2x-2xH+ 在同一个溶胶中, 每个固体微粒所含分子个数 m及其所吸附的离子个数 n 都不一定是相等的. 但整个胶团是电中性的, 总电荷数应为零, 书写胶团结构时要注意这一点")
44
扩散层重叠, 平衡破坏, 产生渗透性斥力和静电斥力
二.溶胶的经典稳定理论——DLVO理论 溶胶是热力学不稳定系统. 但有些溶胶却能在相当长时间里稳定存在.例如法拉弟制备的金溶胶静置数十年才聚沉于管壁上. 为此人们提出了多种理论进行解释, 如DLVO理论, 空间稳定理论及空缺稳定理论等. 1941年由德查金(Darjaguin)和朗道(Landau)以及1948年维韦(Verwey)和奥弗比克(Overbeek)分别提出了带电胶体粒子稳定的理论, 简称DLVO理论. 要点如下: (1) 在胶团之间, 既存在着斥力势能, 又存在着引力势能. 扩散层未重叠, 两胶团之间不产生斥力 扩散层重叠, 平衡破坏, 产生渗透性斥力和静电斥力 00-8-1
和朗道(Landau)以及1948年维韦(Verwey)和奥弗比克(Overbeek)分别提出了带电胶体粒子稳定的理论, 简称DLVO理论. 要点如下: (1) 在胶团之间, 既存在着斥力势能, 又存在着引力势能. 扩散层未重叠, 两胶团之间不产生斥力. 扩散层重叠, 平衡破坏, 产生渗透性斥力和静电斥力")
45
溶胶的经典稳定理论——DLVO理论 两胶团扩散层重叠后, 破坏了扩散层中反离子的平衡分布, 使重叠区反离子向未重叠区扩散, 导致渗透性斥力产生; 同时也破坏了双电层的静电平衡, 导致静电斥力产生. 在溶胶中分散相微粒间存在的吸引力本质上仍具有范德华吸引力的性质, 但这种吸引力的作用范围要比一般分子的大千百倍之多, 故称其为远程范德华力. 远程范德华力势能与粒子间距离的一次方或二次方成正比, 也可能是其它更复杂的关系. (2) 胶体系统的相对稳定或聚沉取决于斥力势能和吸力势能的相对大小. 当粒子间斥力势能在数值上大于吸力势能, 而且足以阻止由于布朗运动使粒子相互碰撞而粘结时, 则胶体处于相对稳定状态; 当吸力势能在数值上大于斥力势能, 粒子将相互靠拢而发生聚沉. 调整两者的相对大小, 可以改变胶体系统的稳定性. 00-8-1
 胶体系统的相对稳定或聚沉取决于斥力势能和吸力势能的相对大小. 当粒子间斥力势能在数值上大于吸力势能, 而且足以阻止由于布朗运动使粒子相互碰撞而粘结时, 则胶体处于相对稳定状态; 当吸力势能在数值上大于斥力势能, 粒子将相互靠拢而发生聚沉. 调整两者的相对大小, 可以改变胶体系统的稳定性")
46
溶胶的经典稳定理论——DLVO理论 (3) 斥力势能, 吸力势能以及总势能都随粒子间距离的变化而变化, 且在某一距离范围吸力占优势, 而在另一距离范围斥力占优势. (4) 加入电解质对吸力势能影响不大, 但对斥力势能的影响却十分显著. 电解质的加入会导致系统的总势能发生很大的变化, 适当调整电解质浓度, 可以得到相对稳定的胶体. 在电荷作用下稳定存在的Fe2O3溶胶 00-8-1
 加入电解质对吸力势能影响不大, 但对斥力势能的影响却十分显著. 电解质的加入会导致系统的总势能发生很大的变化, 适当调整电解质浓度, 可以得到相对稳定的胶体. 在电荷作用下稳定存在的Fe2O3溶胶")
47
溶胶的经典稳定理论——DLVO理论 以粒子间斥力势能ER, 吸力势能 EA 和总势能 E(= ER+EA) 对粒子间距离 x 作图, 得到如图所示的势能曲线. 当 x 缩小, 先出现一极小值a, 则发生粒子的聚集称为絮凝(可逆). x 继续缩小, 则出现极大值Emax(势垒).一般粒子的热运动无法克服它, 使溶胶处于相对稳定状态. 当两胶粒通过热运动积聚的动能超过15kT时才有可能超过此能量值, 进而出现极小值 b, 在此处发生粒子间的聚沉(永久性). 斥力势能, 吸力势能及总势能曲线 00-8-1
.一般粒子的热运动无法克服它, 使溶胶处于相对稳定状态. 当两胶粒通过热运动积聚的动能超过15kT时才有可能超过此能量值, 进而出现极小值 b, 在此处发生粒子间的聚沉(永久性). 斥力势能, 吸力势能及总势能曲线")
48
溶胶的经典稳定理论——DLVO理论 除胶粒带电外, 溶剂化作用也是使溶胶稳定的重要原因. 水化外壳的存在势必增加溶胶聚合的机械阻力.
如前所述, 分散相粒子的布朗运动是胶体粒子受重力影响而不下沉的原因. 当布朗运动足够强时, 粒子热运动能够克服重力场的作用而不下沉, 溶胶的这种性质, 称为动力稳定性. 综上所述, 分散相粒子的带电, 溶剂化作用及布朗运动是溶胶三个重要的稳定原因. 00-8-1

49
三.溶胶的聚沉 聚沉: 憎液溶胶中分散相微粒互相聚结构, 颗粒变大, 进而发生沉淀的现象. 加热, 辐射或加入电解质皆可导致溶胶的聚沉.
三种溶胶聚沉. (左)Al(OH)3; (中)Fe(OH)3; (右)Cu(OH)2 00-8-1
Al(OH)3; (中)Fe(OH)3; (右)Cu(OH)")
50
溶胶的聚沉 (1)电解质的聚沉作用 少量电解质的存在对溶胶起稳定作用; 过量的电解质的存在对溶胶起破坏作用(聚沉). 原因主要是:
电解质的浓度或价数增加都会压缩扩散层, 使扩散层变薄, 斥力势能降低; 若加入的反离子发生特性吸附时, 斯特恩层内的反离子数量增加, 使胶体粒子带电量降低. c1 c2 c3 E x 电解质浓度对胶粒势能的影响 吸力 斥力 从电解质对胶体粒子势能的影响看, 当电解质的浓度或价数增加使溶胶发生聚沉时, 所必须克服的势垒高度和位置皆发生变化, 势垒高度随电解质浓度增大而降低. 00-8-1

51
溶胶的聚沉 聚沉值: 使溶胶发生明显的聚沉所需电解质的最小浓度. 聚沉能力: 聚沉值的倒数定义 为聚沉能力.
舒尔采(Schulze)-哈迪(Hardy)价数规则: 电解质中能使溶胶发生聚沉的离子是反离子, 反离子的价数愈高, 聚沉能力愈强. 如对带负电的As2S3 溶胶起聚沉作用的是电解质的阳离子, KCl, MgCl2, AlCl3的聚沉值分别为49.5, 0.7, 0.093molm-3, 若以K+为比较标准, 其聚沉值有如下关系: Me+ : Me2+ : Me3+ = 1 : 70.7 : 532 一般可近似表示为反离子价数的 6 次方之比, 即 Me+ : Me2+ : Me3+ = 16 : 26 : 36 = 1 : 64 : 729 也有许多反常现象, 如H+虽为一价, 却有很强的聚沉能力. 00-8-1
-哈迪(Hardy)价数规则: 电解质中能使溶胶发生聚沉的离子是反离子, 反离子的价数愈高, 聚沉能力愈强. 如对带负电的As2S3 溶胶起聚沉作用的是电解质的阳离子, KCl, MgCl2, AlCl3的聚沉值分别为49.5, 0.7, 0.093molm-3, 若以K+为比较标准, 其聚沉值有如下关系: Me+ : Me2+ : Me3+ = 1 : 70.7 : 532. 一般可近似表示为反离子价数的 6 次方之比, 即. Me+ : Me2+ : Me3+ = 16 : 26 : 36 = 1 : 64 : 729. 也有许多反常现象, 如H+虽为一价, 却有很强的聚沉能力")
52
溶胶的聚沉 同价离子的聚沉能力也各不相同:
同价正离子, 由于正离子水化能力很强, 且离子半径愈小, 水化能力愈强, 水化层愈厚, 被吸附的能力愈小, 使其进入斯特恩层的数量减少, 而使聚沉值增大. 同价负离子, 由于负离子水化能力很弱, 所以负离子的半径愈小, 吸附能力愈强, 聚沉值愈小. 感胶离子序 将相同电荷的离子按聚沉能力大小排列的顺序。 H+>Cs+>Rb+>NH4+>K+>Na+>Li+ Ba 2+ > Sr 2+ >Ca 2+ >Mg 2+ F->IO3- > H2PO4-> BrO3- > Cl->ClO3-> Br->NO3->ClO4- > I->SCN->OH- 00-8-1

53
溶胶的聚沉 (2)高分子化合物的聚沉作用 高分子化合物既可使溶胶稳定, 也可使溶胶聚沉. 好的聚沉剂应是相对分子量很大的线型聚合物, 如聚丙烯酰胺及其衍生物, 其相对分子量可高达几百万. 聚沉剂可以是离子型的, 也可以是非离子型的. 搭桥效应: 一个长碳链的高聚物分子可以同时吸附在许多个分散相微粒上, 通过“搭桥”把胶粒联结在一起, 引起聚沉. 脱水效应: 高聚物分子由于亲水作用强, 其溶解与水化作用使胶粒脱水, 失去水化外壳而聚沉. 电中和效应: 离子型高聚物吸附在带电的胶粒上而中和了胶粒的表面电荷, 使粒子间的斥力势能降低, 而使溶胶聚沉. 00-8-1

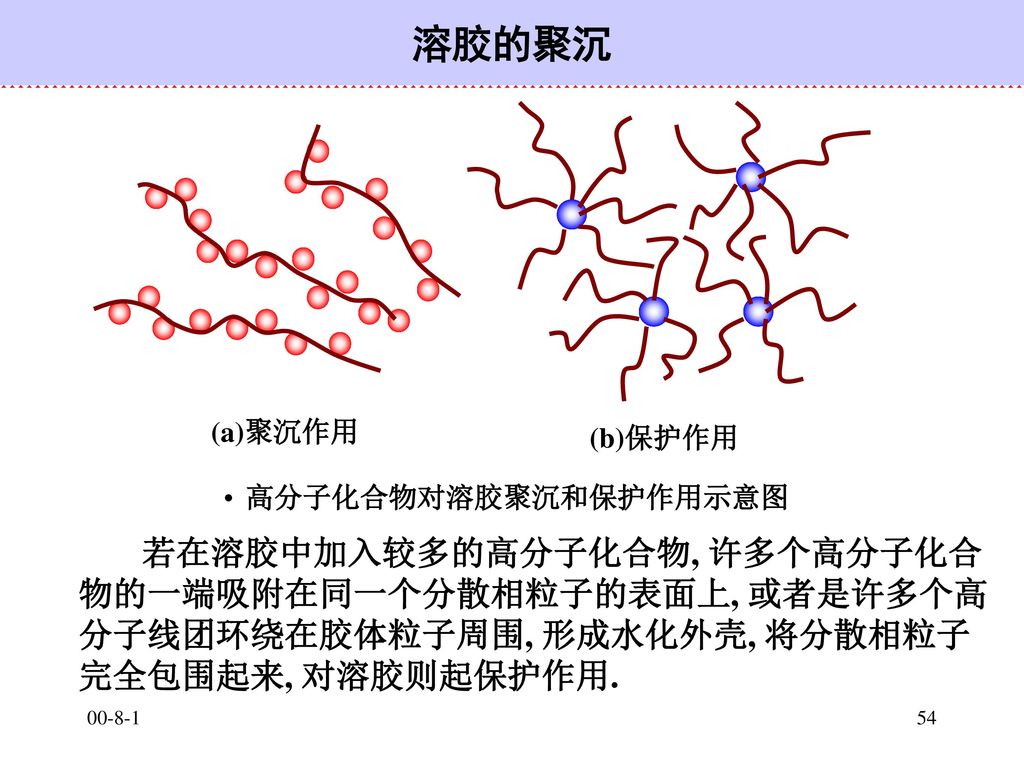
54
溶胶的聚沉 高分子化合物对溶胶聚沉和保护作用示意图 (a)聚沉作用 (b)保护作用 若在溶胶中加入较多的高分子化合物, 许多个高分子化合物的一端吸附在同一个分散相粒子的表面上, 或者是许多个高分子线团环绕在胶体粒子周围, 形成水化外壳, 将分散相粒子完全包围起来, 对溶胶则起保护作用. 00-8-1
55
第七节乳状液、泡沫和气溶胶 一.乳状液 1.乳状液: 由两种不互溶或部分互溶的液体所形成的粗分散系统. 如牛奶, 含水石油, 炼油厂的废水, 乳化农药等. 2.乳化作用: 油水互不相溶, 只有加入乳化剂才能得到比较稳定的乳状液, 乳化剂的这种作用称为乳化作用. 常用的乳化剂多为表面活性剂, 某些固体也能起乳化作用. 3.乳状液的类型:水包油型, 微小油滴分散在水中, 符号O/W. 油包水型, 微小水滴分散在油中, 符号W/O. 复乳型.微小水滴分散在油中,再分散在水中W/O/W 界面膜有两个界面,存在σ水和σ油两个界面张力,此二界面张力大小不同,膜总是向界面张力大的那面弯曲,因这样可减少这个面的面积,使体系趋于稳定,结果在界面张力大的一边的液体就成了分散相。 00-8-1

56
4.乳状液的鉴别 在药物中的应用:缓释,靶向,增溶提高稳定性等 W/O, O/W可增溶,靶向,避免 肠胃液的破坏,方便服用
W/O/W型又称复乳,二级乳,可肠用药(缓释) 内外均带药,既具吸收的迅速,又可持久给药. 鉴别乳状液的类型的方法: 染色法 在乳状液中加入少许油溶性或水溶性的染料, 在显微镜下观察是内相还是外相被染色. 稀释法 取少量乳状液滴入水中或油中, 若乳状液能在水中稀释即为O/W型; 在油中稀释则为W/O型. 导电法 未加离子型乳化剂时, O/W型导电性比W/O强. 00-8-1
 内外均带药,既具吸收的迅速,又可持久给药. 鉴别乳状液的类型的方法: 染色法 在乳状液中加入少许油溶性或水溶性的染料, 在显微镜下观察是内相还是外相被染色. 稀释法 取少量乳状液滴入水中或油中, 若乳状液能在水中稀释即为O/W型; 在油中稀释则为W/O型. 导电法 未加离子型乳化剂时, O/W型导电性比W/O强")
57
5.乳状液的稳定 乳化剂主要从以下几个方面增强乳化液的稳定性. (1) 降低界面张力
乳化液中存在大面积的液-液界面. 加入少量表面活性剂在两相界面产生正吸附, 能显著降低的液-液界面的界面张力, 使系统的表面吉布斯函数降低, 稳定性增加. 另外, 长碳链表面活性剂吸附在两相之间的界面上, 形成一定机械强度的乳化剂膜, 也使得乳状液稳定性增加. 乳化剂膜称为界面相(F), 它与其两边的油和水的界面张力分别以γF-O 及γF-W 表示, 界面总是朝着界面张力大的一方弯曲以使该界面面积较小. 若γF-O > γF-W , 则形成O/W型乳化剂, 一价碱金属皂类易溶于水难溶于油, 属于此类; 若γF-W > γF-O , 则形成W/O型乳化剂, 高价金属皂类易溶于油难溶于水, 属于此类. 00-8-1
, 它与其两边的油和水的界面张力分别以γF-O 及γF-W 表示, 界面总是朝着界面张力大的一方弯曲以使该界面面积较小. 若γF-O > γF-W , 则形成O/W型乳化剂, 一价碱金属皂类易溶于水难溶于油, 属于此类; 若γF-W > γF-O , 则形成W/O型乳化剂, 高价金属皂类易溶于油难溶于水, 属于此类")
58
5.乳状液的稳定 (2)形成定向楔的界面 乳化剂的亲水端和憎水端的截面积常大小不等. 它吸附在油-水界面上时, 常呈现“大头”朝外, “小头”朝里的构型, 使分散相液滴的面积最小, 界面吉布斯函数最低, 且界面膜更牢固. “大头”朝外形成两种类型的乳状液 若亲水基是“大头”, 则亲水基朝外形成水包油型乳化液, 如K, Na等碱金属皂类. 但一价的银肥皂例外. 若憎水基是“大头”, 则憎水基朝外形成油包水型乳化液, 如Ca, Mg, Zn等两价金属皂类. 00-8-1

59
5.乳状液的稳定 (3)形成扩散双电层 乳化剂负离子定向吸附在油-水界面上, 带电的一端指向水, 反离子则呈扩散状分布, 形成扩散双电层, 它一般具有较大的热力学电势及较厚的双电层, 使乳状液处于较稳定的状态. (4)界面膜的稳定作用 乳化过程也是分散相液滴表面的成膜过程, 界面膜的厚度, 特别是强度和韧性, 对乳状液的稳定性起着举足轻重的作用. (5)固体粉末的稳定作用 (左) >90, 颗粒不能被水润湿而更多地进入油中; (中) = 90,颗粒的亲水亲油性均等; (右) < 90, 颗粒能被水润湿而更多地进入水中. 油 水 固体颗粒在油-水界面上的三种润湿情况 00-8-1
界面膜的稳定作用. 乳化过程也是分散相液滴表面的成膜过程, 界面膜的厚度, 特别是强度和韧性, 对乳状液的稳定性起着举足轻重的作用. (5)固体粉末的稳定作用. (左) >90, 颗粒不能被水润湿而更多地进入油中; (中) = 90,颗粒的亲水亲油性均等; (右) < 90, 颗粒能被水润湿而更多地进入水中. 油. 水. 固体颗粒在油-水界面上的三种润湿情况")
60
5.乳状液的稳定 根据空间效应, 为使固体微粒在分散相的周围排列成紧密的固体膜, 固体粒子的大部分应当处在分散介质中, 这样粒子在油-水界面上的不同润湿情况就会产生不同类型的乳状液. 油 水 固体粉末乳化作用示意图 当粒子能被水润湿时, 粒子大部分处于水中, 形成水包油型乳状液, 如粘土, Al2O3等固体微粒; 当粒子易被油润湿时, 粒子大部分处于油中, 形成油包水型乳状液, 如炭黑, 石墨粉等. 固体颗粒的尺寸应远小于分散相的尺寸. 固体表面愈粗糙, 形状愈不对称, 愈易于形成牢固的固体膜, 使乳状液愈稳定. 00-8-1

61
6.乳状液的去乳化 破乳或去乳化作用: 使乳状液破坏的过程. 破乳过程:
分散相的微小液滴首先絮凝成团, 但这时仍未完全失去原来各自的独立性; 分散相凝聚成更大的液滴, 在重力场作用下自动分层. 破乳方法: 用不能形成牢固膜的表面活性物质代替原来的乳化剂; 加入某些能与乳化剂发生化学反应的物质, 消除乳化剂的保护作用. 加入类型相反的乳化剂也可达到破乳的目的 此外, 加热, 加入高价电解质, 加强搅拌, 离心分离, 以及电泳法等皆可加速分散相的聚结, 达到破乳的目的. 00-8-1

62
泡沫 泡沫: 气体分散在液体或固体中所形成的分散系统. 许多大小不等, 形状各异的小气泡堆积在一起, 气泡之间的隔膜各处厚薄不一, 气泡的线度一般在100nm以上, 肉眼可见. 固体泡沫: 分散介质为熔融体, 由于它具有很高粘度, 使小气泡既不易破裂又难于相互靠近, 降温凝固后得到的气-固相分散物质. 如浮石, 泡沫玻璃, 泡沫塑料等 液体泡沫: 分散介质是液体. 表面活性剂的起泡作用 要得到比较稳定的液体泡沫须加入起泡剂, 它们在气-液界面上发生正吸附, 形成定向排列的吸附膜, 能显著地降低气-液界面张力, 又可增加界面膜的机械强度. 00-8-1

63
泡沫 泡沫的应用很广泛, 如泡沫浮选, 泡沫灭火剂, 泡沫杀虫剂, 泡沫除尘及泡沫陶瓷等方面皆用到泡沫技术.
先将矿石粉碎成尺寸在0.1mm以下的颗粒, 加入足量的水、适量的浮选剂和少量起泡剂, 再强烈鼓入空气, 形成大量气泡. 这时憎水性强的矿物附着在气泡上浮至液面, 而被水润湿的长石、石英等沉入水底. 加入浮选剂的作用是增强矿物的憎水性. 泡沫浮选示意图 但在发酵, 精馏, 造纸, 印染及污水处理等工艺过程中, 泡沫的出现将会给操作带来诸多不便, 因此在这类工艺操作中, 必须设法防止泡沫的出现或破坏泡沫的存在. 00-8-1

64
粉尘的分类 气溶胶: 以固体或液体为分散相, 气体为分散介质所形成的胶体系统. 如烟, 雾, 其线度一般在 10nm ~ 1m的数量级, 分散程度高. 而粉尘的分散程度较低, 其线度一般在1m ~ 1mm的数量级, 属粗分散系统, 但性质上与气溶胶有相似之处, 也列入本节讨论. 按粉尘在静止空气中的沉降性质分类: 尘埃: 直径10 ~ 100 m, 颗粒较大, 在静止空气中加速沉降; 尘雾: 直径0. 25 ~ 10 m, 在静空气中可等速沉降; 尘云: 直径 0. 1 m以下, 在静止空气中不能下沉, 而是处于无规则布朗运动状态的浮尘. 00-8-1

65
粉尘的性质 润湿性: 粉尘被水润湿的情况与粉尘的化学性质, 颗粒大小, 带电情况, 温度及接触时间等因素有关. 一般来说, 粉尘颗粒愈小, 吸附性愈强, 它吸附空气中物质粒子形成的气膜愈牢固, 水对其润湿性就愈差. 影响水对粉尘润湿的另一原因是浮于空气中的粉尘质量很小, 遇到净化水幕的雾滴时易产生环绕作用, 不易与水滴接触. 粉尘沉降的速度: 与粉尘颗粒的大小, 形状, 密度等因素有关. 对于分散程度较高, 在静止空气中呈等速沉降的尘粒, 可用斯托克斯方程计算其沉降速度. 实际的大气是不可能处于静止状态的, 粉尘的沉降速度比在静止空气中更慢, 那些半径小于1 m的粉尘, 将长时间地在大气中随风飘移而不下沉. 粉尘的凝聚性: 粉尘经相互碰撞可使微小粉尘聚结成较大的粒子, 当其质量足够大时, 即使有空气干扰, 也能自动沉降. 00-8-1

66
粉尘的性质 粉尘的荷电性: 在粉尘产生过程中, 由于物料之间激烈地磨擦, 撞击, 放射性射线的照射及高压电场的作用, 可使粉尘带电. 粉尘若带有异种电荷, 粒子间吸力增大而易于聚结沉降. 反之则不利于沉降. 带电的粉尘更易于粘附于人的支气管和肺泡上, 对人类产生更大的危害. 粉尘的爆炸性: 粉尘是高度分散的不稳定系统, 在适当条件下就会发生燃烧或爆炸现象. 如镁或碳化钙的粉尘与水接触后会引起燃烧或爆炸. 对这类粉尘不能采用湿式净化设备除尘. 粉尘在空气中的爆炸, 实质上是激烈的化学反应, 只有在一定浓度范围内才可能发生. 发生爆炸时粉尘的最高浓度称为粉尘爆炸上限, 最低浓度称为爆炸下限. 气溶胶的光学性质: 气溶胶的乳光效应基本上也服从雷利公式, 即散射光的强度与入射光波长的4次方成正比. 通过气溶胶的透射光呈橙红色, 散射光为淡蓝色. 00-8-1

67
气体除尘 气体除尘对净化空气和回收有用物质有重大意义.
目前国际上普遍采用静电除尘器, 其除尘效率可达99%. 当含尘气体流经高压静电场时, 在由阴极射出的高能电子束作用下, 气体电离, 并使粉尘带负电荷, 在库仑力作用下射向阳极表面而放电沉积. 某些气溶胶, 如电解NaCl时产生的氢气中含有对人体危害性很大的Hg, 就不宜采用静电降尘的方法. 可采用过硫酸盐溶液对含有Hg的氢气进行化学处理, 处理后Hg蒸气的浓度可降至5 10-6 gm-3以下. 00-8-1

68
悬 浮 液 悬浮液(或悬浮体): 将不溶性固体粒子分散在液体中所形成的粗分散系统.分散相粒子线度大于1000nm, 比憎液溶胶分散相粒子大得多, 因此不存在布朗运动, 不可能产生扩散及渗透现象, 而易于沉降析出. 悬浮液的散射光强度也十分微弱. 粒度分布: 大多数悬浮液都是大小不等的粒子所构成的多级分散系统, 在生产及科研中需了解粒子大小的分布情况. 测定粒度分布的最常用方法是沉降分析. 沉降时, 重力 = (4/3)r3( - 0)g, 阻力 = 6 r. 匀速时两者相等, 可推导出表示沉降速度 的斯托克斯(Stokes)方程: 若在时间 t 内测出粒子的沉降高度 h, = h/t , 得 00-8-1
r3( - 0)g, 阻力 = 6 r. 匀速时两者相等, 可推导出表示沉降速度 的斯托克斯(Stokes)方程: 若在时间 t 内测出粒子的沉降高度 h, = h/t , 得")
69
悬 浮 液 上式表明, 沉降速度与粒子半径 r 的平方成正比! 采用沉降天平可以测出不同时刻 t 的沉降量P, 可得沉降曲线.
(a)粒子单级分散 (b)粒子二级分散 (c)粒子多级分散 沉降曲线 作某时刻的P- t 线切线, 求得切线在纵坐标上的截距. 然后由前式求出此时刻对应的粒子半径r, 截距的值即为半径大于r 的粒子全部沉降的量. 此值与所有粒子的完全沉降量之比即为半径大于此r的粒子占全部粒子的质量分数Q. 作Q - r 线即得到粒子的积分分布曲线. 00-8-1
粒子单级分散. (b)粒子二级分散. (c)粒子多级分散. 沉降曲线. 作某时刻的P- t 线切线, 求得切线在纵坐标上的截距. 然后由前式求出此时刻对应的粒子半径r, 截距的值即为半径大于r 的粒子全部沉降的量. 此值与所有粒子的完全沉降量之比即为半径大于此r的粒子占全部粒子的质量分数Q. 作Q - r 线即得到粒子的积分分布曲线")
70
悬 浮 液 r Q Q - r 曲线 r dQ / dr - r 曲线 dQ / dr 由Q - r 曲线进一步作dQ/dr - r 线, 即得到粒子的微分分布曲线, 此曲线表示了不同半径范围的粒子占全部粒子的质量分数. 沉降分析常用于测定悬浮液的粒度分布, 在许多领域如土壤学, 硅酸盐, 颜料等的生产和科研中有着广泛的应用. 00-8-1

71
盐析作用 对于高分子溶液来说, 加入少量电解质, 它的稳定性并不会受到影响, 到了等电点也不会聚沉, 直到加入更多电解质, 才能使它发生聚沉. 高分子溶液的这种聚沉现象称为盐析. 发生盐析作用的主要原因是去水化. 有些高分子化合物含有可以电离的极性基从而使分子带电, 少量电解质的加入可以引起动电电势的降低, 但并不能使它失去稳定性, 这时分子仍是高度水化的, 只有加入更多的盐才会出现盐析现象. 实验表明, 盐析能力与盐类离子的种类有关. 一些离子盐析能力的顺序(也称感胶离子序)是: (阴)柠檬酸>酒石酸>SO42->醋酸>Cl->NO3->ClO2->I-. (阳) Li+>K+>Na+>NH4+>Mg2+ 这种顺序与离子的水化顺序极为一致. 00-8-1
是: (阴)柠檬酸>酒石酸>SO42->醋酸>Cl->NO3->ClO2->I-. (阳) Li+>K+>Na+>NH4+>Mg2+ 这种顺序与离子的水化顺序极为一致")
72
胶凝现象, 触变现象和脱水收缩 胶凝现象: 高分子溶液在适当条件下可以失去流动性, 整个系统变为弹性半固体状态, 这种系统叫做凝胶; 液体含量较多的凝胶也叫做胶冻. 高分子溶液(或溶胶)形成凝胶的过程叫做胶凝作用. 触变现象: 有些凝胶的网状结构不稳定, 可因机械力变成有较大流动性的溶液状态, 外力解除静置后又恢复凝胶状态, 这种现象叫做触变. 脱水收缩: 胶凝作用并非凝聚的终点, 在许多情况下, 如将凝胶放置时, 就开始渗出微小的液滴, 这些液滴逐渐合并而形成一个液相, 与此同时凝胶本身的体积将缩小, 且乳光度亦随之增加. 这种使凝胶分成两相的作用称为脱水收缩. 00-8-1

73
胶凝现象, 触变现象和脱水收缩 A colloid. When gelatin is mixed with boiling water, it forms a colloidal dispersion called a sol. When cooled, the dispersion is called a gel. 00-8-1

74
凝胶的溶胀 溶胀: 有的凝胶失去水分或重新吸收分散介质后, 形状和体积几乎不变, 这种凝胶为脆性凝胶; 有的凝胶当失去分散介质后, 体积显著缩小, 但当重新吸收分散介质时, 体积又重新膨胀, 这种凝胶为弹性溶胶. 干燥的弹性凝胶吸收分散介质而体积增大的现象称为溶胀. 无限溶胀: 溶胀是高分子化合物溶解的第一阶段. 对于某些物质在一定溶剂中,最后达到全部溶解, 称为无限溶胀. 有限溶胀: 另一些高分子化合物, 由于形成了有交联的网状结构, 在溶胀过程中, 所吸收的液体量达到最大值, 而不再继续膨胀, 这种溶胀现象称为有限溶胀. 00-8-1

75
高分子溶液与憎液溶胶的区别 高分子化合物: 摩尔质量 M > 1~ 104kgmol-1的大分子化合物, 它们在适当的溶剂中, 可自动地分散成溶液, 称为高分子溶液. 高分子化合物是以分子或离子状态均匀地分布在溶液中, 在分散质与分散介质之间无相界面存在. 故高分子溶液是均匀分布的真溶液, 即热力学平衡系统. 这是高分子溶液与憎液溶胶的最本质的区别. 由于高分子化合物分子的大小恰好是在胶体范围内, 而且又具有胶体系统的某些特性, 如扩散速度慢, 不能通过半透膜, 在超级离心机中可进行沉降分离. 因此又将高分子溶液称为亲液溶胶. 00-8-1

76
高分子溶液与憎液溶胶的区别 性质 憎液溶胶 高分子溶液 高分子溶液与溶胶性质的对比 粒子大小 1100nm 分散质存在形式
若干分子形成的胶粒 单个分子 能否透过半透膜 不能 扩散速度 慢 系统性质 多相、热力学 不稳定系统 均相、热力学 稳定系统 丁铎尔效应 强 微弱 粘度大小 小(与纯溶剂粘度相似) 大 对电解质的 敏感性 敏感(加入少量电解质就会聚沉) 不敏感(加入大量电解质会发生盐析) 干燥或聚沉后 能否复原 能 00-8-1
 大. 对电解质的. 敏感性. 敏感(加入少量电解质就会聚沉) 不敏感(加入大量电解质会发生盐析) 干燥或聚沉后. 能否复原. 能")
77
高分子溶液的渗透压 非电解质稀溶液或理想稀溶液的渗透压公式为
在高分子溶液中, 分散质与介质之间存在着较强的亲合力, 产生明显的溶剂化效应, 这势必影响溶液的渗透压. 若以 B 代表溶质的质量浓度(SI单位kgm-3), M 为溶质的质量摩尔质量, 则上式可改写为 实验表明, 在恒温下 /B并不是一个常数, 而是随B的变化而变化. 在这种情况下, 可采用维里方程的模型来表示渗透压 1 与质量浓度 B 之间的关系, 即 式中常数A2, A3, …称为维里系数. 若浓度很小, 上式可简化为 00-8-1
, M 为溶质的质量摩尔质量, 则上式可改写为. 实验表明, 在恒温下 /B并不是一个常数, 而是随B的变化而变化. 在这种情况下, 可采用维里方程的模型来表示渗透压 1 与质量浓度 B 之间的关系, 即. 式中常数A2, A3, …称为维里系数. 若浓度很小, 上式可简化为")
78
高分子溶液的渗透压 在恒温下, 若以 /B对B 作图, 应得一直线, 可由该直线的斜率及截距计算高分子化合物的摩尔质量M和第二维里系数A2. 渗透压法测定高分子摩尔质量的范围是10 ~103kgmol-1, 摩尔质量太小时, 容易通过半透膜, 制膜有困难; 太大时渗透压很低, 测量误差大. 上式只适用于不能电离的高分子化合物. 对于蛋白质水溶液, 只有在等电点时才能适用, 对于可电离的高分子化合物, 上式求出的摩尔质量往往偏低, 这主要是电解质电离的影响. 唐南(Donnan)提出的离子隔膜平衡理论, 令人满意地解释了许多这类实验的情况. 00-8-1
提出的离子隔膜平衡理论, 令人满意地解释了许多这类实验的情况")
79
渗透平衡时NaCl在两相中的化学势相等. 由化学势表达式 (NaCl) = (NaCl) + ln{c(Na+) c(Cl-)}
唐南平衡 以 Na+z Pz-代表蛋白质, 其电离反应可以表示为 每个分子电离产生(z + 1)个离子. 用半透膜将纯水与蛋白质水溶液隔开, Pz-不能通过半透膜. 对应的渗透压力可表示为 如果另一侧不是纯水而是电解质水溶液, 则有如下过程发生: Na+, Pz- zc, c Na+, Cl- c, c Na+, Pz-, Cl- zc + x, c, x Na+, Cl- c -x, c -x 始态 平衡态 左 右 唐南平衡示意图 渗透平衡时NaCl在两相中的化学势相等. 由化学势表达式 (NaCl) = (NaCl) + ln{c(Na+) c(Cl-)} 00-8-1
个离子. 用半透膜将纯水与蛋白质水溶液隔开, Pz-不能通过半透膜. 对应的渗透压力可表示为. 如果另一侧不是纯水而是电解质水溶液, 则有如下过程发生: Na+, Pz- zc, c. Na+, Cl- c, c Na+, Pz-, Cl- zc + x, c, x. Na+, Cl- c -x, c -x. 始态. 平衡态. 左. 右. 唐南平衡示意图. 渗透平衡时NaCl在两相中的化学势相等. 由化学势表达式. (NaCl) = (NaCl) + ln{c(Na+) c(Cl-)}")
80
唐南平衡 c(Na+, 左) c(Cl- , 左) = c(Na+, 右) c(Cl- , 右)
(zc + x) x = (c - x)2 求得 x = c 2 / (2 c + zc) 唐南平衡: 半透膜两侧不仅溶液成平衡, 电解质也达到平衡. 唐南平衡最重要的功能是控制物质的渗透压, 这对医学和生物学等研究细胞内外的渗透平衡有重要意义. 00-8-1
 x = (c - x)2. 求得 x = c 2 / (2 c + zc) 唐南平衡: 半透膜两侧不仅溶液成平衡, 电解质也达到平衡. 唐南平衡最重要的功能是控制物质的渗透压, 这对医学和生物学等研究细胞内外的渗透平衡有重要意义")
81
高分子溶液的粘度 当液体流动时, 液体内部的分子会产生摩擦力, 阻碍液体的相对流动. 以层流为例:
流体在管道中呈层流流动时流速的径向分布 y x 当液体流动时, 液体内部的分子会产生摩擦力, 阻碍液体的相对流动. 以层流为例: 可把液体分成许多层, 吸附在管壁上的一层液体是不动的, 而管中心的流速为最大, 这种流型称为牛顿型. 设相邻两平行流层面积为As, 层间距为dx, 流速差为d, 则两层间的流速梯度d /dx, 即为切变速率. 在稳态流动时, 推动液体流动的外力F, 在数值上应等于液体流动时所产生的摩擦力, 即 牛顿粘性流动定律: 式中 为摩擦系数, 即动力粘度, 简称粘度. 上式可改写为 00-8-1

82
高分子溶液的粘度 式中 称为剪切应力. 在一定温度下 为常数. 几种粘度定义: 00-8-1

83
高分子溶液的粘度 高分子溶液的粘度较一般憎液溶胶或普通溶液的粘度大得多. 如, 若在苯中溶入质量百分数为1%的橡胶, 该溶液粘度要比纯苯的粘度大十多倍. 如图所示, 当高分子溶液的浓度增加时, 其粘度急剧上升. 此外, 高分子溶液的粘度还与溶质的大小, 形状及溶剂化程度等因素有关. 最常用的高分子溶液的粘度与相对分子质量关系的经验方程为 粘度 分散质浓度 高分子溶液 憎液溶胶 溶胶浓度对粘度的影响 00-8-1

84
高分子溶液的粘度 , K为系统的特征参数, 它们的大小取决于溶质及溶剂的性质. 主要取决于高分子在溶液中的形态, 若分子卷曲成线团状, < 1而近于0.5; 当线团松解成弯曲的线状时, = 1; 若高分子链伸直为棍状, 则 = 2. 实验表明, K的大小不仅与溶液性质有关, 而且还随溶质的相对分子质量的增加而降低. 用粘度法测定高分子的相对质量时, 必须先用其它方法, 例如光散射法, 直接测出系统的 及K. 再测出在不同浓度c下相应的增比粘度sp, 然后以sp / c 对c 作图, 可得一直线, 将直线外推至 c = 0 处, 所得截距即为 [] . 除少数蛋白质外, 高分子化合物都是由相对分子质量大小不等, 结构也不完全相同的同系混合物. 因此, 不论用什么方法测得的分子量都是在一定范围内的平均值. 同一个高分子溶液, 用不同方法测得的平均分子量往往具有不同的名称和不同的数值. 由粘度法测定的分子量则称为粘均分子量. 00-8-1


 如何区分元素和微粒(分子、原子、离子) 1 .概念的范畴不同 元素 —— 宏观概念, 只论种类不论个数 微粒 —— 微观概念, 既论种类又论个数 2 .决定种类的因素不同 元素 —— 由核电荷数 ( 即质子数 ) 决定 (如 16.>")
 A. 四氧化三铁 B. 河水 C. 稀盐酸 D. 高锰酸钾制氧气后的剩余物 2. 有浓盐酸、硫酸铜、氯化铁三种溶液,可 以把它们区别开的性质是( ) A. 状态 B. 气味 C. 颜色 D. 以上三者都可区别.>")






 提出离子键理论。 一 离子键的形成>")










