Download presentation
1
第2章 药物的体内过程

2
药物的体内过程:从药物进入机体至排出体外的过程
ADME四大体内过程:吸收、分布、代谢(生物转化)、排泄。 吸收、分布与排泄 机体对药物的转运。
、排泄。 吸收、分布与排泄 机体对药物的转运。")
3
第1节 药物的转运机制与转运体

4
药物的转运机制 被动转运 载体转运 滤过 简单扩散 易化扩散 主动转运

5
药物的转运 组织器官 Free Bound 吸收 游离型药 排泄 SYSTEMIC CIRCULATION 结合型药 生物转化

6
脂质双分子层

7
一、 被动转运 又名“下山”或顺流转运,它是指药物依赖于膜两侧的浓度差从高浓度一侧通过通过物理扩散过程经生物膜向低浓度一侧的转运过程。
一、 被动转运 又名“下山”或顺流转运,它是指药物依赖于膜两侧的浓度差从高浓度一侧通过通过物理扩散过程经生物膜向低浓度一侧的转运过程。 特点: (1)不消耗能量 (2)无饱和现象 (3)无竞争性抑制
不消耗能量. (2)无饱和现象. (3)无竞争性抑制.")
8
(一)滤过(filtration) 药物通过亲水膜孔的转运 在流体静压或渗透压的作用下,许多小的、水溶性的极性物质和非极性物质的转运方式
滤过(filtration) 药物通过亲水膜孔的转运 在流体静压或渗透压的作用下,许多小的、水溶性的极性物质和非极性物质的转运方式")
9
(二)简单扩散(simple diffusion)
是药物转运的一种最常见、最重要的形式 速度取决于 脂溶性越大(油水分布系数越大)、浓度梯度越高,扩散就越快。脂溶性与解离度有关。 膜两侧的药物浓度梯度 药物的脂溶性
、浓度梯度越高,扩散就越快。脂溶性与解离度有关。 膜两侧的药物浓度梯度. 药物的脂溶性.")
10
酸性药 (Acidic drug): HA H+ + A 碱性药 (Basic drug): BH+ H+ + B (分子型)
离子障(ion trapping) 非解离型极性低,亲脂,可通过膜;解离型相反
 非解离型极性低,亲脂,可通过膜;解离型相反.")
11
弱酸性和弱碱性药物的pKa 酸性药 : 碱性药: Ka = pKa = pH - log 10 pH-pKa = [ H+ ] [ A ]
[ BH+ ] [B] 碱性药: 10 pKa-pH =
![弱酸性和弱碱性药物的pKa 酸性药 : 碱性药: Ka = pKa = pH - log 10 pH-pKa = [ H+ ] [ A ]](http://slidesplayer.com/slide/11251446/61/images/11/%E5%BC%B1%E9%85%B8%E6%80%A7%E5%92%8C%E5%BC%B1%E7%A2%B1%E6%80%A7%E8%8D%AF%E7%89%A9%E7%9A%84pKa+%E9%85%B8%E6%80%A7%E8%8D%AF+%EF%BC%9A+%E7%A2%B1%E6%80%A7%E8%8D%AF%EF%BC%9A+Ka+%3D+pKa+%3D+pH+-+log+10+pH-pKa+%3D+%5B+H%2B+%5D+%5B+A%EF%80%AD+%5D.jpg "[ BH+ ] [B] 碱性药: 10 pKa-pH =")
12
色甘酸钠 :pka=2,弱酸性 pH=4 pH=7 HAH+ + A A + H+HA 1 102 105 1
总量 101 HAH+ + A A + H+HA 总量 100001 1 102 105 1 弱酸性药物的解离随着pH的升高而增加, 弱碱性药物的解离随着pH的升高而减少。 [ A ] [HA] [ A ] [HA] 10pH-pKa = 10pH-pKa = = 104-2 = 102 = 107-2 = 105

13
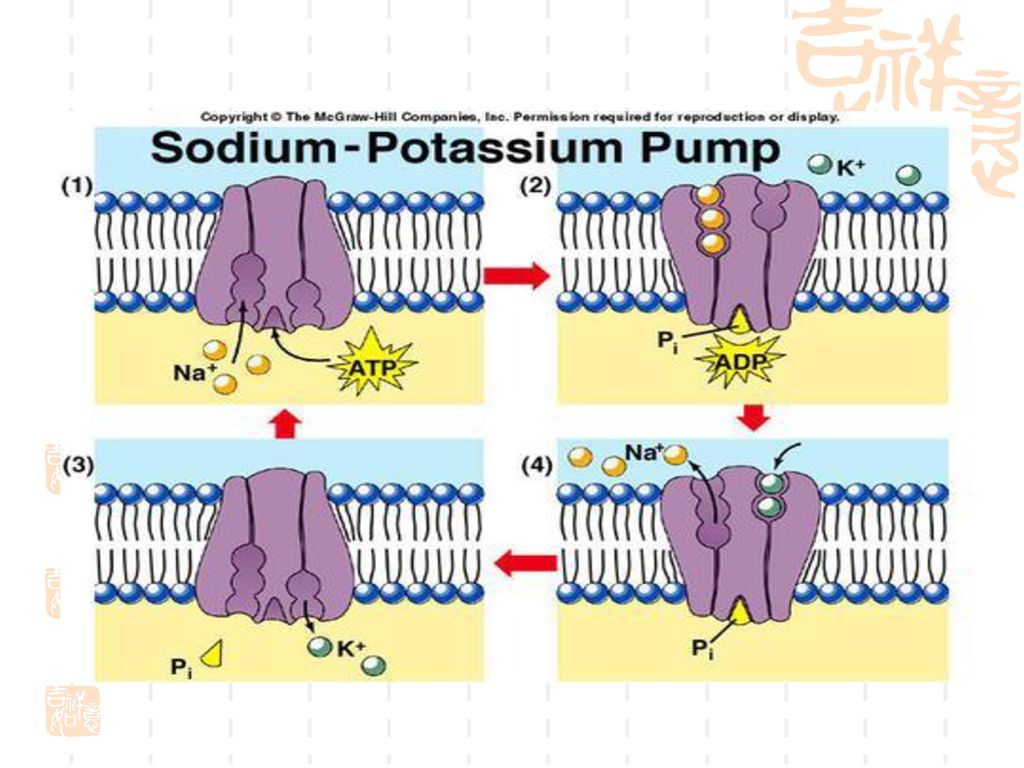
二、载体转运 (一)主动转运:“上山”或逆流转运。药物从低浓度一侧经生物膜向高浓度一侧的转运过程。 特点: 1.需要载体 2.消耗细胞能量
3.有饱和现象 4.有竞争性抑制

15
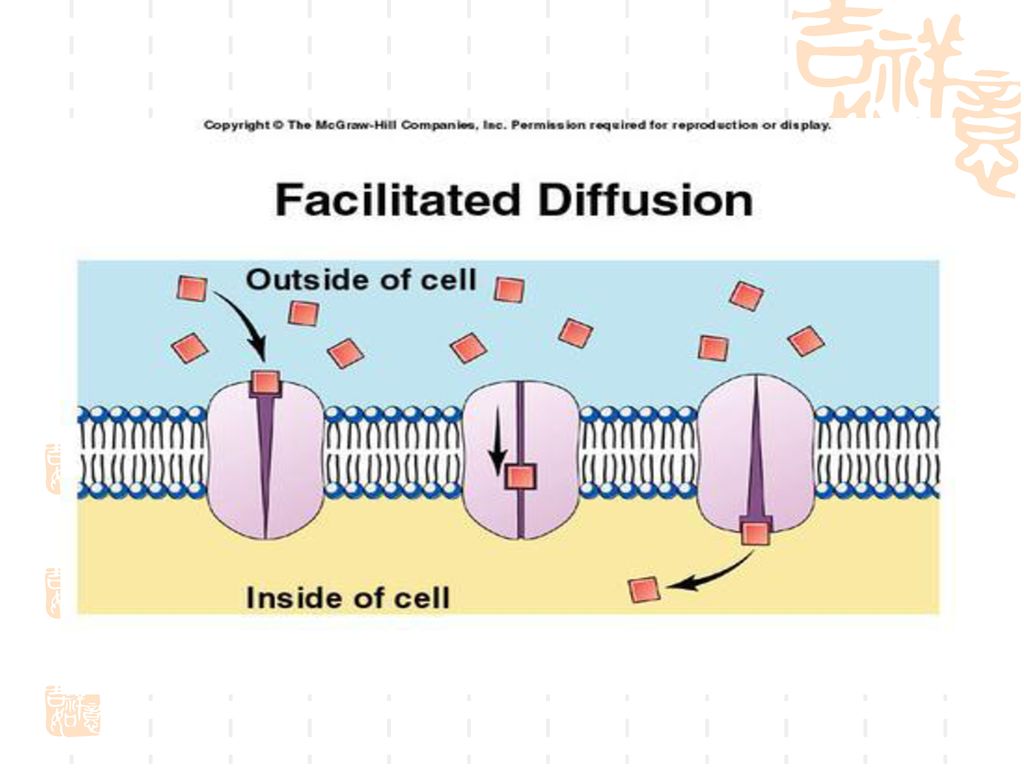
(二)易化扩散 特点: 不耗能(因为不逆浓度梯度) 有饱和现象 有竞争性抑制(因为需要载体)
易化扩散 特点: 不耗能(因为不逆浓度梯度) 有饱和现象 有竞争性抑制(因为需要载体)")
17
转运的形式:

18
二、药物转运体 药物的体内转运过程,包括吸收、分布、代谢和排泄过程都涉及药物对细胞膜的转运。对于细胞膜对药物的通透性,以往主要从药物的理化性质,如亲脂性和水溶性的方面研究比较多。 近年来研究发现,在某些情况下,增加药物的脂溶性,并不一定能增加细胞膜对药物的通透性。而进一步研究表明,许多组织和器官的生物膜存在着一种特殊的转运蛋白系统介导药物跨膜转运,称为转运体 ( transporters)。
。")
19
二、药物转运体 常见的药物转运体 药物转运体按其转运的方向不同大致可分为两种。一种是摄取型转运体,可转运药物进入细胞,增加细胞内药物浓度,比如:有机阴离子多肽转运体 (OATP)、有机阳离子转运体 (OCT)和寡肽转运体 (PEPT)等都属此类转运体;另一种是外排型转运体,依赖ATP分解释放的能量,将药物逆向转运出细胞,降低药物在细胞内的浓度,比如乳腺癌耐药蛋白 (BCRP)、肺耐药蛋白(LRP)、多药抗性相关蛋白 (MRP)、P糖蛋白 (Pgp)等转运体都属于这种类型。
、有机阳离子转运体 (OCT)和寡肽转运体 (PEPT)等都属此类转运体;另一种是外排型转运体,依赖ATP分解释放的能量,将药物逆向转运出细胞,降低药物在细胞内的浓度,比如乳腺癌耐药蛋白 (BCRP)、肺耐药蛋白(LRP)、多药抗性相关蛋白 (MRP)、P糖蛋白 (Pgp)等转运体都属于这种类型。")
20
二、药物转运体 P-糖蛋白(P-gp) 1976年首先在多药耐药(MDR)癌细胞系中发现高表达的P-gp,并发现P-gp水平的高低与细胞膜的通透性、细胞内药物浓度以及耐药程度有着密切的关系。P-gp是一种重要的主动转运载体,除了在肿瘤细胞中有分布外;在人体正常组织也有分布,且表达水平存在一定的个体差异。P-gp主要位于这些细胞的绒毛膜表面的一侧。P-gp有逆向转运药物的功能,降低细胞内药物的浓度。
癌细胞系中发现高表达的P-gp,并发现P-gp水平的高低与细胞膜的通透性、细胞内药物浓度以及耐药程度有着密切的关系。P-gp是一种重要的主动转运载体,除了在肿瘤细胞中有分布外;在人体正常组织也有分布,且表达水平存在一定的个体差异。P-gp主要位于这些细胞的绒毛膜表面的一侧。P-gp有逆向转运药物的功能,降低细胞内药物的浓度。")
21
二、药物转运体 药物转运体对体内过程的影响 吸收
由于转运体存在多态性,导致有关药物在吸收速率方面存在较大变异,并成为口服药物吸收非线性机制的原因。转运体在药物吸收中具有泵入与泵出的作用。例如,P-糖蛋白还限制了许多药物的吸收,例如环孢素、地高辛和他林洛尔等。而且,P-糖蛋白在人类消化道的各个部分发挥的作用是不同的。从口腔开始往下,各部分的P-糖蛋白水平逐段增高,因此药物在不同部位的吸收程度就不相同,沿着胃肠道往下血药浓度逐渐降低。

22
二、药物转运体 药物转运体对体内过程的影响 分布
体内的一些屏障组织大都存在P-糖蛋白等转运体,也能将药物外排到细胞外,从而改变药物在局部组织的分布。过去人们认为,增加药物的亲脂性可以提高它们通过血脑血障的通透性。但后来发现,有些的脂溶性很高,但通过血脑屏障的能力却很低,这主要是因为大脑中的毛细血管内皮细胞膜上存在转运体,可将这些药物从内皮细胞内部泵回到血液。

23
二、药物转运体 药物转运体对体内过程的影响 分布
胎盘屏障中P-糖蛋白的逆向转运,可以降低胎儿与药物的接触。因此,孕期内要慎用含有P-糖蛋白抑制剂的药物,以降低药物对胎儿的损害。 药物在房水、晶状体和玻璃体等组织的浓度远低于血液这是由血眼屏障的作用所致,转运体在这方面也起重要的作用。抑制血眼屏障转运体对药物的逆向转运可提高药物在眼中的浓度。故作用于眼的药物多采用局部用药的方式。

24
二、药物转运体 药物转运体对体内过程的影响 代谢
肝药酶与P-糖蛋白在小肠表皮细胞中相邻存在。通常,药物经口服后通过被动扩散进入小肠表皮细胞,在此可被肝药酶代谢,或被P-糖蛋白转运回到肠腔。 它们还常具有共同的诱导剂或抑制剂,因而肝药酶与P-糖蛋白之存在着相互作用的关系。

25
二、药物转运体 药物转运体对体内过程的影响 排泄 P-糖蛋白在肾脏主要分布于近端小管上皮细胞;参与肾脏的药物排泄。
药物经肝排入胆汁,转运体也发挥着重要作用。

26
第2节 吸 收 指药物未经化学变化而进入血流的过程。
通常认为,只有吸收的药物,才能发挥预期疗效,因此,药物吸收的多少与难易,对药物作用有决定性的影响。

27
消化道内吸收 消化道外吸收 口腔吸收(舌下给药:硝酸甘油) 从胃、小肠、直肠吸收 从注射部位吸收(静脉注射和静脉滴注) 从皮肤黏膜吸收
从鼻黏膜、支气管或肺泡吸收

28
静脉内给药无吸收过程 其它给药途径按吸收速度排序: 吸入→舌下→直肠→肌注→皮下→口服→皮肤

29
影响药物从消化道内吸收的主要因素 (一)药物自身因素 药物的pKa和脂溶性对药物吸收有着明显的影响,在药剂学的研究中,对胃肠道不易吸收的药物,将其分子结构稍加改变,则可使吸收量增加,例如将红霉素制成丙酸酯,口服后可使红霉素的血药浓度增高数倍,这是因为形成红霉素丙酸酯后,脂/水分配系数增大,同时pKa的数值也有所改变所致。

30
1.胃肠pH 2.胃排空速度和肠蠕动 (二)机体因素 抑制胃酸分泌的药物如阿托品、普鲁本辛及抗酸药能碱化胃内容物,将使弱酸行药物在胃吸收减少
小肠是药物吸收的主要部位(因有大量的绒毛及其上面的微绒毛增加了有效的吸收面积) 2.胃排空速度和肠蠕动 延缓胃排空的因素:食物中含脂肪酸或脂肪、高浓度的电解质或氢离子、高粘度食物 加速胃排空的因素:进食、饥饿、甲亢等。
 2.胃排空速度和肠蠕动. 延缓胃排空的因素:食物中含脂肪酸或脂肪、高浓度的电解质或氢离子、高粘度食物. 加速胃排空的因素:进食、饥饿、甲亢等。")
31
3.胃肠食物及其他内容物 可使药物吸收减少或者增加。在进餐时服用四环素、林可霉素、利福平等药物则明显的降低了药物的吸收。核黄素饱餐时服用,药物的吸收率反而比空腹时的吸收率高。灰黄霉素则会因服用脂肪餐而使吸收量加倍。

32
首关效应(first-pass effect)
首关效应是指某些药物首次通过肠壁或经门静脉进入肝脏时被其中的酶所代谢致使进入人体循环药量减少的一种现象。具有明显首关消除的药物有:阿司匹林、氯丙嗪、异丙肾上腺素、硝酸甘油等。 硝酸甘油的首关消除可灭活90%,口服疗效差,改为舌下含服。

33
消化道外吸收 静脉注射 (intravenous injection) 药物直接进入血液循环,无吸收过程,起效快
药物不宜口服、皮下或肌内注射,需迅速发生药效时,可采用静脉注射法。 输液和输血,以及用于静脉营养治疗。

34
呼吸道吸入给药 (Inhalation) 气体或挥发性液体麻醉药和其他气雾剂型药物可通过呼吸道吸收。
肺有很大表面积,血流量大,经肺的血流量约为全身的10%,肺泡细胞结构较薄,故药物极易吸收。

35
第3节 分 布 分布是指吸收入血的药物随血流转运至组织器官的过程。大部分药物的分布过程属于被动转运,少数为主动转运。包括两部分:
药物在血液中的分布 药物在血液与组织间的分布

36
一、药物在血液中的分布 (一)与血细胞结合 50%~60%的环孢素与红细胞结合 米帕林在白细胞的浓度高于血浆浓度100倍
(二)与血浆蛋白结合 与血浆蛋白结合的药物称结合型药物(bound drug) 未与血浆蛋白结合的药物称游离型药物(free drug)
与血浆蛋白结合. 与血浆蛋白结合的药物称结合型药物(bound drug) 未与血浆蛋白结合的药物称游离型药物(free drug)")
37
血浆蛋白结合率 药物与血浆蛋白结合的程度常以结合药物的浓度与总浓度比值表示,称为血浆蛋白结合率。 ①酸性药物主要与白蛋白结合; ②碱性药物主要与α1酸性糖蛋白或脂蛋白结合; ③许多内源性物质及维生素等主要与球蛋白结合。

38
血浆中结合型药物百分比 乙琥胺 0 阿莫西林 18 地高辛 25 吗啡 35 茶碱 65 甲氧苄啶 70 氨苯蝶啶 81 苯妥英 91
药物 结合型药物百分比 乙琥胺 阿莫西林 地高辛 吗啡 茶碱 甲氧苄啶 氨苯蝶啶 苯妥英 阿米替林 氯贝丁酯 甲苯磺丁脲 呋塞米(速尿) 地西泮(安定) 华法林
 98. 地西泮(安定) 98. 华法林 99.")
39
药物与血浆蛋白结合的特点 药物与血浆蛋白借助范德华力、氢键或离子键。 药物与血浆蛋白的结合是可逆的,结合与解离处于动态平衡。
结合型药物限制了跨膜转运,结合型药物常失去药理活性。只有游离型药物具有药理活性。 血浆蛋白的数量是有限的,药物与血浆蛋白的结合也是有一定限度的。

40
影响血浆蛋白结合率的因素 (一)血浆蛋白结合位点
血浆蛋白的量和药物结合部位是有限的,当同时应用一种或多种药物时,可能在蛋白结合部位发生竞争,蛋白结合率高的药物可将结合率低的药物从结合位点上置换下来,使后者游离药物比率增高,提高其药理活性或毒性。(保泰松和华法林)
")
41
(二)疾病的影响 某些疾病(急性病毒性肝炎或肝硬化),能降低许多药物与血浆蛋白的结合率,使血浆中游离型药物浓度增加。
血浆白蛋白、1-酸性糖蛋白合成降低; 血浆蛋白结合部位减少; 内源性抑制物蓄积,抑制物能与药物竞争血浆蛋白结合部位,从而降低了药物与血浆蛋白的结合。

42
二、药物在血液与组织间的分布 (一)体液pH值
细胞内液pH=7.0,细胞外液pH=7.4,解救弱酸性药物苯巴比妥中毒用NaHCO3,碱化血液和尿液,加速转运和排泄。 (二)器官血流量与膜的通透性 肝、肾、脑、肺等高灌注器官,药物分布较快 肌肉、皮肤等,药物分布较慢 (三)组织细胞结合 碘在甲状腺中的浓度比其他组织约高1万倍 氯喹在肝内浓度比血浆浓度高700倍,治疗阿 米巴性肝脓肿
器官血流量与膜的通透性. 肝、肾、脑、肺等高灌注器官,药物分布较快. 肌肉、皮肤等,药物分布较慢. (三)组织细胞结合. 碘在甲状腺中的浓度比其他组织约高1万倍. 氯喹在肝内浓度比血浆浓度高700倍,治疗阿 米巴性肝脓肿.")
43
(四)体内屏障 1.血脑屏障(blood-brain barrier,BBB)
体内屏障 1.血脑屏障(blood-brain barrier,BBB)")
44
某些大分子、水溶性或解离型药物难于进入脑组织;
有机酸或碱性药物进入脑组织缓慢; 而乙醚、硫喷妥等脂溶性很高的药物,则能迅速向脑内转运,血液中浓度与脑内浓度几乎瞬间达到平衡,这些药物向脑内的转运仅与进入脑内的血流量有关。

45
2.胎盘屏障(placental barrier)
脂溶性药物能以简单扩散的方式经胎盘而进入胎儿体内,水溶性或高解离度的药物则不易通透。由于有些药物对胎儿毒性较大,并可导致畸形,因此孕妇用药应特别慎重。

46
第4节 代 谢 药物进入体内后,发生化学结构上的变化,这就是药物代谢过程,也可称为生物转化。肝脏是最主要的代谢器官。
第4节 代 谢 药物进入体内后,发生化学结构上的变化,这就是药物代谢过程,也可称为生物转化。肝脏是最主要的代谢器官。 药物被代谢后:①多数可能转化为无活性物质;②也可能从原来无药理活性的物质转变为有活性的代谢物;③甚至有时可能生成有毒物质。

47
生物转化的步骤: I相反应 氧化、还原、水解:是向底物分子引入极性基团如-OH、-COOH、-NH2、 -SHO
(1)提高分子的极性,即水溶性 (2)为第二步代谢做准备 Ⅱ相反应 结合:将在第一步代谢中引入的底物分子极性基团,分别与葡萄糖醛酸、甘氨酸、硫酸结合,形成水溶性更大的复合物,随尿、胆汁排出。
提高分子的极性,即水溶性. (2)为第二步代谢做准备. Ⅱ相反应 结合:将在第一步代谢中引入的底物分子极性基团,分别与葡萄糖醛酸、甘氨酸、硫酸结合,形成水溶性更大的复合物,随尿、胆汁排出。")
48
生物转化酶的分类 1.专一性酶:胆碱酯酶、儿茶酚氧位甲基转 移酶(COMT)、单胺氧化酶(MAO)等
2.非专一性酶:细胞色素P-450(Cytochrome P-450)简称“肝药酶”,通常肝药酶位于线粒体、细胞核胞膜、内质网。
简称 肝药酶 ,通常肝药酶位于线粒体、细胞核胞膜、内质网。")
49
P-450催化反应的机制: 2.接受还原型NADPH的一个电子,形成还原型P450-药物复合物(Fe2+)
3+4:复合物结合一分子氧后,在接受一个电子使氧气活化为氧离子。 5.活化的氧离子将药物氧化,并与两个质子生成水

50
生物转化的差异性及其影响因素 (一)遗传因素 存在种属、种族和个体差异 不同种属酶水解的速度是:小鼠=田鼠>大鼠>狗>猴>人
遗传因素 存在种属、种族和个体差异 不同种属酶水解的速度是:小鼠=田鼠>大鼠>狗>猴>人")
51
(二)环境因素 许多药物对肝药酶具有诱导或抑制作用,直接关系到药物的清除速率,改变药物作用的持续时间与强度。
①诱导剂:某些化学物质能提高肝药酶活性,增加自身或其他药物的代谢速率。包括苯巴比妥和其他巴比妥类药物、苯妥英钠、卡马西平、利福平、水合氯醛等。共同特点是:亲脂、易与细胞色素P450结合并具有较长的半衰期。 ②抑制剂:能抑制肝药酶活性,减慢其他药物的代谢速率。包括氯霉素、对氨基水杨酸、异烟肼和保泰松等。药物代谢的抑制常与抑制剂的血药浓度有关。

52
生物转化的差异性及其影响因素 (三)食物与营养状态 (四)年龄与性别 (五)病理因素
食物与营养状态 (四)年龄与性别 (五)病理因素")
53
第5节 排 泄 排泄是指体内药物或其代谢物排出体外的过程。 器官:肾脏、胆汁、肠、肺、乳腺、唾液或汗腺排出。

54
1.肾排泄 肾脏是最重要的排泄器官

55
(一)肾小球滤过 肾小球滤过膜呈筛状,筛孔较大,除了与血浆蛋白结合的药物外,游离药物及药物的代谢物均通过肾小球滤过进入肾小管。影响肾小球药物滤过的主要因素是肾小球滤过率与药物血浆蛋白结合程度。
肾小球滤过 肾小球滤过膜呈筛状,筛孔较大,除了与血浆蛋白结合的药物外,游离药物及药物的代谢物均通过肾小球滤过进入肾小管。影响肾小球药物滤过的主要因素是肾小球滤过率与药物血浆蛋白结合程度。")
56
(二)肾小管分泌 肾小管分泌主要在近曲小管进行,是主动转运过程,需要载体参与,有饱和现象,不受蛋白结合率影响。转运载体包括有机酸转运载体和有机碱转运载体,分别分泌有机酸类药物和有机碱类药物,如果分泌机制相同的两种药物合用,可发生竞争性抑制(丙磺舒和青霉素)。
肾小管分泌 肾小管分泌主要在近曲小管进行,是主动转运过程,需要载体参与,有饱和现象,不受蛋白结合率影响。转运载体包括有机酸转运载体和有机碱转运载体,分别分泌有机酸类药物和有机碱类药物,如果分泌机制相同的两种药物合用,可发生竞争性抑制(丙磺舒和青霉素)。")
57
(三)肾小管的重吸收 肾小管是脂类屏障,重吸收主要是简单扩散和主动转运。
脂溶性大的药物易被再吸收,排泄缓慢;尿液pH影响药物重吸收。碱化尿液使酸性药物在尿中离子化,酸化尿液使碱性药物在尿中离子化,阻止药物重吸收。 肾排泄是肾小球滤过、肾小管重吸收及肾小管分泌的总和。

58
2.胆汁排泄 肝脏至少有三个彼此独立的载体主动转系统,分别如下: ①阴离子(有机酸类如对氨基马尿酸、磺溴酞、青霉素等) ②阳离子(有机碱类如奎宁、红霉素等) ③中性化合物如强心苷等 肝脏排泌有机酸和有机碱至胆汁的机制也存在同类药物相互竞争的现象

59
肝肠循环(hepatoenteral circulation)
药物与葡萄糖醛酸等结合后排入胆汁,随胆汁到达小肠后被水解,游离药物被重吸收。 肝肠循环可以延长药物的作用时间。















>")





