Download presentation
Presentation is loading. Please wait.

1
第10章 生物信息学基础

2
1简介 5蛋白质数据分析 6 分子进化分析 2 常用的数据库 contents 3数据库检索 7 多态性分析 4 核酸数据分析

3
1 简介

4
生物信息学(bioinformatics)是80年代未随着人类基因组计划(Human genome project)的启动而兴起的一门新的交叉学科。它涉及生物学、数学、计算机科学和工程学,并依赖于生物实验和衍生数据的大量储存。 利用计算机运算法则和计算机的数据库分析 proteins, genes 和 genomes,使用生物信息学工具的目的是读懂测序得到的上亿个DNA碱基对。 Sanger与合作者发现(Sanger, 1952),胰岛素蛋白质包含了固定组成的氨基酸。之前的观点认为蛋白质可能是由相似氨基酸堆积在一起的混合物,而不具有特定的结构。而Sanger发现胰岛素蛋白质作为一种纯净物(单一的分子),并且具有特定的三级结构。因此,Sanger推断蛋白质是排列完美的分子,而这种排列的完美性,其中应当蕴含着一些未知的机理。 Sanger与合作者分别对牛、猪和羊的胰岛素蛋白质进行了测序(Brown et al., 1955; Ryle et al., 1955),并做了序列上的比较。 1962年鲍林提出分子进化的理论(Pauling, 1964),这为接下来的生物信息学的发展,尤其是基于序列比对来发现同源/相似蛋白质的计算工作奠定了初步的理论基础。鲍林推测在人中,可能会存在大约50,000~100,000个不同的蛋白质,而由于当时已经知道基因编码蛋白质,且当时的理论认为一个基因编码一个蛋白质,鲍林推测人中可能会有大约50,000~100,000个不同的基因。鲍林的理论只对了一半。目前已知的人的基因是21,037个(Imanishi et al., 2004),虽然可能以后的实验会继续发现遗漏的基因,但预测的结果表明大约是~30,000左右,鲍林的推测显然是不符合的。而鲍林认为人中可能存在大约50,000~100,000个不同的蛋白质的推测,则是正确的。究其原因,是基因在翻译的时候存在可变剪切(Alternatively Splicing, AS)的现象,一个基因可以翻译成多种不同的蛋白质。 1970年,Needleman-Wunsch,全局优化的序列比对算法:允许匹配、错配和缺失。动态规划的算法:任务可分割,分成更小的子问题进行解决。 1981年,Smith-Waterman,局部优化的序列比对算法。
,胰岛素蛋白质包含了固定组成的氨基酸。之前的观点认为蛋白质可能是由相似氨基酸堆积在一起的混合物,而不具有特定的结构。而Sanger发现胰岛素蛋白质作为一种纯净物(单一的分子),并且具有特定的三级结构。因此,Sanger推断蛋白质是排列完美的分子,而这种排列的完美性,其中应当蕴含着一些未知的机理。 Sanger与合作者分别对牛、猪和羊的胰岛素蛋白质进行了测序(Brown et al., 1955; Ryle et al., 1955),并做了序列上的比较。 1962年鲍林提出分子进化的理论(Pauling, 1964),这为接下来的生物信息学的发展,尤其是基于序列比对来发现同源/相似蛋白质的计算工作奠定了初步的理论基础。鲍林推测在人中,可能会存在大约50,000~100,000个不同的蛋白质,而由于当时已经知道基因编码蛋白质,且当时的理论认为一个基因编码一个蛋白质,鲍林推测人中可能会有大约50,000~100,000个不同的基因。鲍林的理论只对了一半。目前已知的人的基因是21,037个(Imanishi et al., 2004),虽然可能以后的实验会继续发现遗漏的基因,但预测的结果表明大约是~30,000左右,鲍林的推测显然是不符合的。而鲍林认为人中可能存在大约50,000~100,000个不同的蛋白质的推测,则是正确的。究其原因,是基因在翻译的时候存在可变剪切(Alternatively Splicing, AS)的现象,一个基因可以翻译成多种不同的蛋白质。 1970年,Needleman-Wunsch,全局优化的序列比对算法:允许匹配、错配和缺失。动态规划的算法:任务可分割,分成更小的子问题进行解决。 1981年,Smith-Waterman,局部优化的序列比对算法。")
5
生物信息学概念的提出 1956年,在美国田纳西州盖特林堡召开的首次“生物学 中的信息理论研讨会”上,产生了生物信息学概念。但 直到20世纪80-90年代,伴随着计算机科学技术的进步, 生物信息学才获得突破性进展。 1987年,马来西亚华裔林华安博士正式把这一学科命名 为“生物信息学”(Bioinformatics)。此后,其内涵 随着研究的深入和现实需要的变化而几经更迭。 1991年,Bioinformatics这个词第一次出现在文献中 1995年,在美国人类基因组计划第一个五年总结报告中 ,给出了一个较为完整的生物信息学定义:生信息学是 包含生物信息的获取、处理、贮存、分发、分析和解释 的所有方面的一门学科,它综合运用数学、计算机科学 和生物学的各种工具进行研究,目的在于了解大量的生 物学意义。 虽然直到1991年才第一次出现在文献中,但生物信息学实际上已经存在了30多年,只不过最初被称为基因组信息学。当然它还有一些其他的名字,如计算生物学、计算分子生物学、生物分子信息学等。
。此后,其内涵 随着研究的深入和现实需要的变化而几经更迭。 1991年,Bioinformatics这个词第一次出现在文献中. 1995年,在美国人类基因组计划第一个五年总结报告中 ,给出了一个较为完整的生物信息学定义:生信息学是 包含生物信息的获取、处理、贮存、分发、分析和解释 的所有方面的一门学科,它综合运用数学、计算机科学 和生物学的各种工具进行研究,目的在于了解大量的生 物学意义。 虽然直到1991年才第一次出现在文献中,但生物信息学实际上已经存在了30多年,只不过最初被称为基因组信息学。当然它还有一些其他的名字,如计算生物学、计算分子生物学、生物分子信息学等。")
6
Sanger与合作者发现(Sanger, 1952),胰岛素蛋白质包含了固定组成的氨基酸。之前的观点认为蛋白质可能是由相似氨基酸堆积在一起的混合物,而不具有特定的结构。而Sanger发现胰岛素蛋白质作为一种纯净物(单一的分子),并且具有特定的三级结构。因此,Sanger推断蛋白质是排列完美的分子,而这种排列的完美性,其中应当蕴含着一些未知的机理。 Sanger与合作者分别对牛、猪和羊的胰岛素蛋白质进行了测序(Brown et al., 1955; Ryle et al., 1955),并做了序列上的比较。 1962年鲍林提出分子进化的理论(Pauling, 1964),这为接下来的生物信息学的发展,尤其是基于序列比对来发现同源/相似蛋白质的计算工作奠定了初步的理论基础。鲍林推测在人中,可能会存在大约50,000~100,000个不同的蛋白质,而由于当时已经知道基因编码蛋白质,且当时的理论认为一个基因编码一个蛋白质,鲍林推测人中可能会有大约50,000~100,000个不同的基因。鲍林的理论只对了一半。目前已知的人的基因是21,037个(Imanishi et al., 2004),虽然可能以后的实验会继续发现遗漏的基因,但预测的结果表明大约是~30,000左右,鲍林的推测显然是不符合的。而鲍林认为人中可能存在大约50,000~100,000个不同的蛋白质的推测,则是正确的。究其原因,是基因在翻译的时候存在可变剪切(Alternatively Splicing, AS)的现象,一个基因可以翻译成多种不同的蛋白质。 1970年,Needleman-Wunsch,全局优化的序列比对算法:允许匹配、错配和缺失。动态规划的算法:任务可分割,分成更小的子问题进行解决。 1981年,Smith-Waterman,局部优化的序列比对算法。
,并做了序列上的比较。 1962年鲍林提出分子进化的理论(Pauling, 1964),这为接下来的生物信息学的发展,尤其是基于序列比对来发现同源/相似蛋白质的计算工作奠定了初步的理论基础。鲍林推测在人中,可能会存在大约50,000~100,000个不同的蛋白质,而由于当时已经知道基因编码蛋白质,且当时的理论认为一个基因编码一个蛋白质,鲍林推测人中可能会有大约50,000~100,000个不同的基因。鲍林的理论只对了一半。目前已知的人的基因是21,037个(Imanishi et al., 2004),虽然可能以后的实验会继续发现遗漏的基因,但预测的结果表明大约是~30,000左右,鲍林的推测显然是不符合的。而鲍林认为人中可能存在大约50,000~100,000个不同的蛋白质的推测,则是正确的。究其原因,是基因在翻译的时候存在可变剪切(Alternatively Splicing, AS)的现象,一个基因可以翻译成多种不同的蛋白质。 1970年,Needleman-Wunsch,全局优化的序列比对算法:允许匹配、错配和缺失。动态规划的算法:任务可分割,分成更小的子问题进行解决。 1981年,Smith-Waterman,局部优化的序列比对算法。")
7
生物信息学? 与分子生物学的发展 海量的生物数据的产生: 1995破译了全长180万bp的嗜血流感杆菌基因组。
生物信息学? 与分子生物学的发展 海量的生物数据的产生: 1995破译了全长180万bp的嗜血流感杆菌基因组。 2001年的春天,公布了人类基因组的工作草图。 1999年12月DNA碱基数目为30亿, 2000年4月DNA碱基数目是60亿 更为本质的原因是基因组数据的复杂性。用 A T G C代表生物的遗传密码,人的遗传密码就含有32亿个字符,构成了一部100多万页、每页有3000字符的“天书”

8
GenBank生物数据

9
国际著名的生物信息中心 NCBI National Center for Biotechnology Information (US)
EBI European Bioinformatics Institute (EU) HGMP Human Genome Mapping Project Resource Centre (UK) ExPASy Expert of Protein Analysis System (Switzerland ) CMBI Centre of Molecular and Biomolecule (The Netherlands) ANGIS National Genome Information Service (Australia) NIG National Institute of Genetics (Japan) BIC National Bioinformatics Centre (Singapore)
 HGMP Human Genome Mapping Project Resource Centre. (UK) ExPASy Expert of Protein Analysis System (Switzerland ) CMBI Centre of Molecular and Biomolecule (The Netherlands) ANGIS National Genome Information Service (Australia) NIG National Institute of Genetics (Japan) BIC National Bioinformatics Centre (Singapore)")
10
生物信息学主要研究内容 生物分子数据的收集与管理 新基因的发现与鉴定 非编码区的信息结构分析 生物进化 的研究 完整基因组的比较
基因组信息分析方法的研究 大规模基因功能表达谱分析 蛋白质三级结构的预测、模拟和分子设计

11
2 常用的数据库

12
生物信息学数据库 一级数据库 二级数据库 蛋白质结构数据库 DNA数据库 基因组数据库 蛋白质序列数据库

13
重要的一级数据库 三大核酸序列数据库 两大蛋白序列数据库 蛋白质结构数据库:PDB 整合后的蛋白质数据库:UniProt数据库
GenBank(美国NCBI) EMBL(欧洲EBI) DDBJ(日本NIG) SWISS-PROT库(瑞士) PIR库(美国) 整合后的蛋白质数据库:UniProt数据库 这三大数据库虽然各自有不同的数据记录格式,但对核酸序列均采用相同的记录标准,同时每天交换数据以达到数据更新和一致。从地域角度看,EMBL主要负责收集欧洲的数据,GenBank负责美洲,DDBJ负责亚洲。由于国际互联网的发展,用户可以任意的向其中任意一个数据库提交序列,所提交的序列也将从公布之日起同时在三大数据库中出现。 蛋白质结构数据库:PDB
 EMBL(欧洲EBI) DDBJ(日本NIG) SWISS-PROT库(瑞士) PIR库(美国) 整合后的蛋白质数据库:UniProt数据库. 这三大数据库虽然各自有不同的数据记录格式,但对核酸序列均采用相同的记录标准,同时每天交换数据以达到数据更新和一致。从地域角度看,EMBL主要负责收集欧洲的数据,GenBank负责美洲,DDBJ负责亚洲。由于国际互联网的发展,用户可以任意的向其中任意一个数据库提交序列,所提交的序列也将从公布之日起同时在三大数据库中出现。 蛋白质结构数据库:PDB.")
14
GenBank(美国国家生物技术信息中心,NCBI)
1982年4月:由以下三个机构联合建立 NIH(National Institute of Health,美国国立卫生研究院) NCBI(National Center for Biotechnology Information,美国国家 生物技术信息中心(做日常维护)) NLM(National Library of Medicine,美国国立医学图书馆) GenBank数据库最初设在美国洛斯阿拉莫斯国家实验室(LANL),现由位于美国马里兰州Bethesda的国家生物技术研究中心(NCBI)维护管理。数据库每日更新,每年发行六版。 其中所收集的序列包括:基因组DNA序列、cDNA序列、EST序列、STS序列、载体序列、人工合成序列及HTG序列等。 GenBank自创建以来就与EMBL核酸序列数据库进行了国际合作。
 NCBI(National Center for Biotechnology Information,美国国家 生物技术信息中心(做日常维护)) NLM(National Library of Medicine,美国国立医学图书馆) GenBank数据库最初设在美国洛斯阿拉莫斯国家实验室(LANL),现由位于美国马里兰州Bethesda的国家生物技术研究中心(NCBI)维护管理。数据库每日更新,每年发行六版。 其中所收集的序列包括:基因组DNA序列、cDNA序列、EST序列、STS序列、载体序列、人工合成序列及HTG序列等。 GenBank自创建以来就与EMBL核酸序列数据库进行了国际合作。")
15
NCBI主页http://www.ncbi.nlm.nih.gov 检索关键词 常用资源 下拉菜单里是各种数据库 常用资源
主页的导航条有七大类: PubMed:上千万条文献记录及许多在线期刊连接; All Databases:NCBI中的各种数据库集合; BLAST:局部比对的序列相似性搜索工具; OMM:在线人类孟德尔遗传性状数据库,人类基因和遗传异常的索引; Books:在线的参考书籍,包含与PubMEd的链接; Taxonomy:囊括主要生物类别的分类信息浏览器; Structure:分子建模数据库,记录了大分子的三维结构 NCBI主页

16
两大蛋白质数据库 SWISS-PROT蛋白质数据库
1986年欧洲瑞士日内瓦大学的Amos Bairoch设计了一个蛋白质序列分析工具(COMPSEQ-PC/Gene)并建立了第一个全新的蛋白质序列数据库SwissProt,该数据库的所有条目都经过有经验的分子生物学家和蛋白质化学家通过计算机工具并查阅有关文献资料仔细核实,因此又称蛋白质专家库(ExPASy)。 可从中搜索,获得各种蛋白质的氨基酸序列,及其各种配基结合位点、酶活位点等。 以蛋白质氨基酸顺序及注释信息为基本内容的数据库称为蛋白质序列数据库。蛋白质序列测定技术的发明早于DNA序列测定技术(1952年Sanger测定了一条蛋白质——胰岛素的序列,1977年Sanger等发明了DNA序列测定技术),蛋白质序列的搜集整理也早于DNA序列,但蛋白质序列数据库建立比核酸序列数据库要晚一些。1984年“蛋白质信息资源”(Protein Informaton Resource,简称PIR)计划正式启动,蛋白质序列数据库PIR-PSD也因此而诞生。1988年日本的国际蛋白质信息数据库和德国的慕尼黑蛋白质序列信息中心加入PIR,合作成立了国际蛋白质信息中心。 (1)从核酸数据库经过翻译推导而来;(2)从蛋白质数据库PIR挑选出合适的数据;(3)从科学文献中摘录;(4)研究人员直接提交的蛋白质序列数据。
并建立了第一个全新的蛋白质序列数据库SwissProt,该数据库的所有条目都经过有经验的分子生物学家和蛋白质化学家通过计算机工具并查阅有关文献资料仔细核实,因此又称蛋白质专家库(ExPASy)。 可从中搜索,获得各种蛋白质的氨基酸序列,及其各种配基结合位点、酶活位点等。 以蛋白质氨基酸顺序及注释信息为基本内容的数据库称为蛋白质序列数据库。蛋白质序列测定技术的发明早于DNA序列测定技术(1952年Sanger测定了一条蛋白质——胰岛素的序列,1977年Sanger等发明了DNA序列测定技术),蛋白质序列的搜集整理也早于DNA序列,但蛋白质序列数据库建立比核酸序列数据库要晚一些。1984年 蛋白质信息资源 (Protein Informaton Resource,简称PIR)计划正式启动,蛋白质序列数据库PIR-PSD也因此而诞生。1988年日本的国际蛋白质信息数据库和德国的慕尼黑蛋白质序列信息中心加入PIR,合作成立了国际蛋白质信息中心。 (1)从核酸数据库经过翻译推导而来;(2)从蛋白质数据库PIR挑选出合适的数据;(3)从科学文献中摘录;(4)研究人员直接提交的蛋白质序列数据。")
17
SwissProt蛋白质序列数据库在国际上比较权威,一般任何蛋白质序列数据搜寻和比较都应从SwissProt开始。
SwissProt涉及已知蛋白质的功能、序列(包括一些蛋白质片断序列)、结构域(如跨膜区等)结构、翻译后修饰(如磷酸化与去磷酸化等)及其位点、突变体等。SwissProt还与其他一些数据库如Prosite、Swiss-2D PAGE、Swiss-3D IMAGE、Enzyme、SwissModel、NCBI等相链接。
、结构域(如跨膜区等)结构、翻译后修饰(如磷酸化与去磷酸化等)及其位点、突变体等。SwissProt还与其他一些数据库如Prosite、Swiss-2D PAGE、Swiss-3D IMAGE、Enzyme、SwissModel、NCBI等相链接。")
18
检索关键词 常用资源 SWISS-PROT蛋白质数据库 主页:

19
UniProt数据库 2002年为了整合全球的蛋白质序列资源,使信息共享,美国的蛋白质信息资源数据库PIR与欧洲生物信息学研究所EBI、瑞士生物信息学研究所SIB在国立卫生研究院NIH的资助下,决定建立全球范围内统一的蛋白质序列和功能数据库UniProt(通用蛋白质资源,Universal Protein Resource)。 合并了分属不同研究所下的PIR-PSD(美国)、SwissProt(瑞士)和 TrEMBL(欧洲)数据库。合并后的蛋白质数据库Uniprot具有全世界最全面的蛋白质分类信息,是蛋白质序列与功能主要的知识库。 UniProt包括三个子库: (1)蛋白质知识库(UniProt Knowledgebase,简称UniProtKB),包含蛋白质序列全面的信息、提供蛋白质准确、丰富的序列与功能注释。 (2)蛋白质参考子集库(UniProt Reference Clusters,简称UniRef),把非常相似的序列整合到一个记录中,形成适合检索的数据子集(reference clusters),提供检索效率。 (3)蛋白质文档库(UniProt Archive,简称UniParc),储存了所有公共数据库中有效的蛋白质序列数据,包括蛋白质序列的来源以及来源数据库的链接。
。 合并了分属不同研究所下的PIR-PSD(美国)、SwissProt(瑞士)和 TrEMBL(欧洲)数据库。合并后的蛋白质数据库Uniprot具有全世界最全面的蛋白质分类信息,是蛋白质序列与功能主要的知识库。 UniProt包括三个子库: (1)蛋白质知识库(UniProt Knowledgebase,简称UniProtKB),包含蛋白质序列全面的信息、提供蛋白质准确、丰富的序列与功能注释。 (2)蛋白质参考子集库(UniProt Reference Clusters,简称UniRef),把非常相似的序列整合到一个记录中,形成适合检索的数据子集(reference clusters),提供检索效率。 (3)蛋白质文档库(UniProt Archive,简称UniParc),储存了所有公共数据库中有效的蛋白质序列数据,包括蛋白质序列的来源以及来源数据库的链接。")
20
UniProt is a collaboration between the European Bioinformatics Institute (EBI), the SIB Swiss Institute of Bioinformatics and the Protein Information Resource (PIR). Across the three institutes more than 100 people are involved through different tasks such as database curation, software development and support.

21
检索关键词 UniProt数据库网址:

22
蛋白质三级结构数据库(PDB) 是国际上唯一的生物大分子结构数据档案库,由美国Brookhaven国家实验室建立。
PDB收集的数据主要来源于X光晶体衍射和核磁共振(NMR)的数据,经过整理和确认后存档而成。目前PDB数据库的维护由结构生物信息学研究合作组织(RCSB)负责。使用Rasmol等软件可以在计算机上按PDB文件显示生物大分子的三维结构。
的数据,经过整理和确认后存档而成。目前PDB数据库的维护由结构生物信息学研究合作组织(RCSB)负责。使用Rasmol等软件可以在计算机上按PDB文件显示生物大分子的三维结构。")
23
检索关键词 网址:

25
PDB Current Holdings Breakdown11
Exp.Method Proteins Nucleic Acids Protein/NA Complexes Other Total X-RAY 73689 1450 3793 2 78934 NMR 8657 1028 192 7 9884 ELECTRON MICROSCOPY 355 41 126 522 HYBRID 46 3 1 52 other 146 4 6 13 169 82893 2526 4119 23 89561

26
基因组数据库 基因组数据库的主体是模式生物基因组数据库,是一个比较专一的数据库,只收录单一的物种序列、结构、发育等相关数据信息,因此也仅对所对应的研究领域及相关研究领域有价值。 来源于人类基因组计划及各种模式生物基因组计划 人类、线虫、拟南芥、家蚕、水稻、家鸡、小鼠、斑马鱼、猪、 马…… 1977年,最早获得的生物基因组全序列是噬菌体(53kb) 1995年,人类测定了第一个能独立存活的生命体的基因组——流感嗜血菌(Haemophilus. influenzae)
 1995年,人类测定了第一个能独立存活的生命体的基因组——流感嗜血菌(Haemophilus. influenzae)")
27
NCBI的“genome”数据库中集成了众多物种的基因组数据
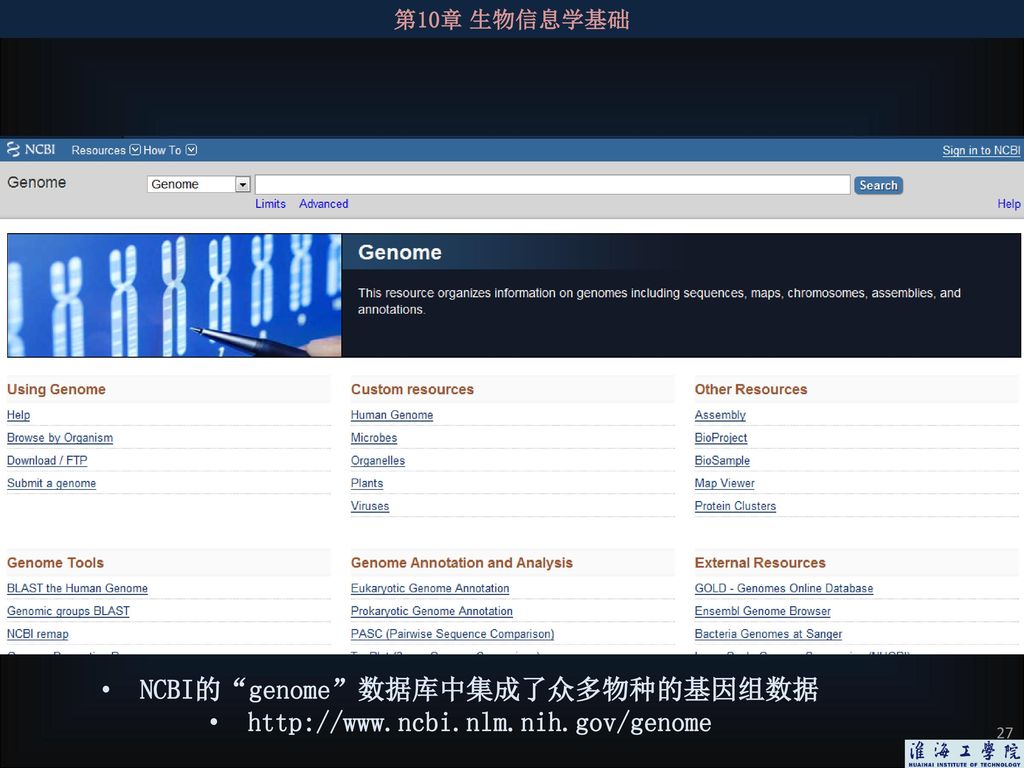
28
3 数据库检索

29
3.1 在Entrez数据库中检索 Entrez系统是NCBI的核心检索系统,管理了NCBI的主要生物信息资源,包括DNA序列数据库、蛋白质序列数据库和UniGene数据库等。Entrez根据用户要求可在多组数据库之间进行检索。 网址:

30
Entrez系统主页: 搜索内容 动作按钮 各数据库链接

31
也可在NCBI主页上直接用关键词搜索 NOT.AND.OR
如:选择“all databases”,搜索关键词“neuraminidase”(H7N9的神经氨酸酶)
")
32
PUBMED收录的文献 不同数据库前的数字表示搜索结果。 核酸 蛋白质 结构 …………

33
有“free”标记的文献可免费下载全文,未标记“feee”的并不表示不能下载到全文
当然还可以用“google学术”、Springer数据库、Elsevier数据库等其他工具进行搜索获取全文。

34
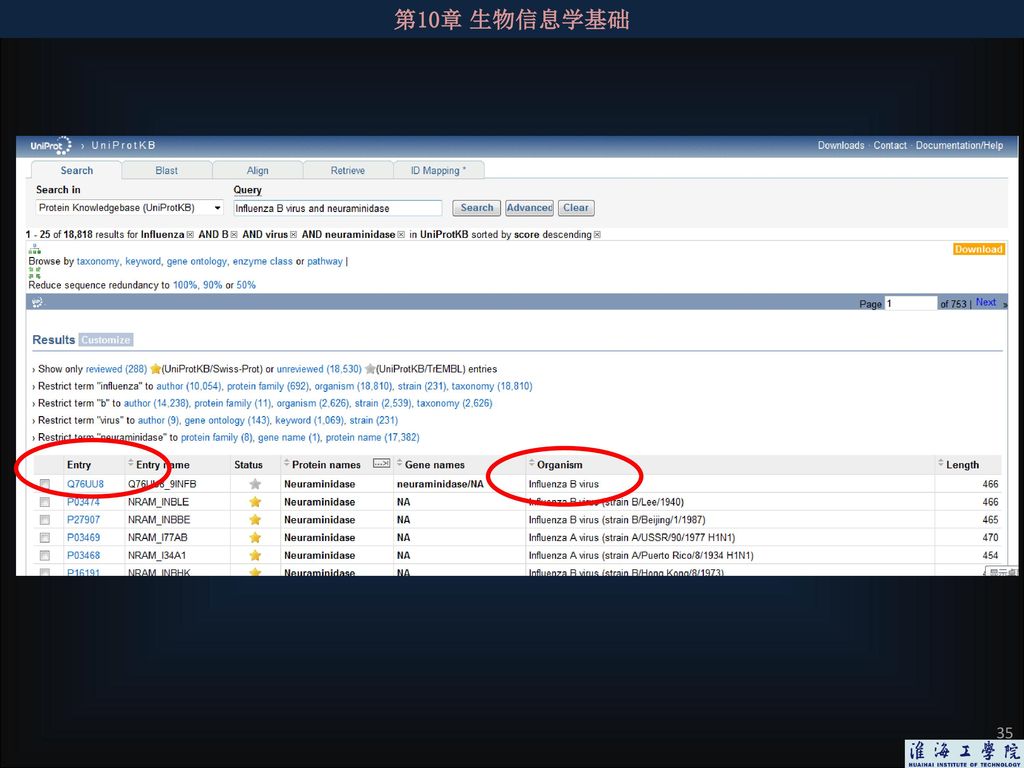
3.2 如何从数据库下载序列 例:要下载乙型流感病毒的神经氨酸酶蛋白质序列,可登陆UniPort网站,输入关键词“neuraminidase”和“Influenza B virus”,以及逻辑联接词“and”

36
这里可以找到这个蛋白质相对应的核酸序列。 无论是下载蛋白质序列还是DNA序列,最好都选择fasta格式下载。

37
Fasta格式 FASTA格式(又称为Pearson格式),是一种基于文本用于表示核苷酸序列或氨基酸序列的格式。在这种格式中碱基对或氨基酸用单个字母来编码,且允许在序列前添加序列名及注释 序列文件的第一行是由大于号“>”或分号“;”打头的任意文字说明(习惯常用“>”作为起始),用于序列标记。从第二行开始为序列本身,只允许使用既定的核苷酸或氨基酸编码符号。通常核苷酸符号大小写均可,而氨基酸常用大写字母。使用时应注意有些程序对大小写有明确要求。文件每行的字母一般不应超过80个字符。 。
,用于序列标记。从第二行开始为序列本身,只允许使用既定的核苷酸或氨基酸编码符号。通常核苷酸符号大小写均可,而氨基酸常用大写字母。使用时应注意有些程序对大小写有明确要求。文件每行的字母一般不应超过80个字符。 。")
38
例如乙型流感病毒的神经氨酸酶基因 起始密码子 终止密码子

39
以Fasta格式下载的核酸序列或蛋白质序列可以用许多软件打开,如DNAStar软件包的EditSeq。
所得序列就是B型流感病毒的“neuraminidase”蛋白序列,可用于各种分析。

40
这里可以找到这个蛋白质相对应的核酸序列。
选择核酸序列正确的链接

41
选择FASTA格式下载 这个就是乙型流感病毒的“neuraminidase”核酸序列的fasta格式,在页面下半部分也能看到,可用于复制保存至文本文件(可将后缀名由.txt改成.fasta)。
。")
42
当然在NCBI主页上,可选择“nucleotide”数据库,并输入关键词“neuraminidase”和“Influenza B virus”,以及逻辑联接词“and”进行搜索。但是出来的数据太多,不好甄别。

43
4 核酸数据分析 对生物序列的研究是现代生命科学当中的一个关键而基本的问题。这里面涉及的内容很多,而核心问题则是比较各种不同类型的生物体序列的相互关系。当许多基因组的测序工作完成之后,寻找物种内和物种间蛋白质的相关性对我们理解生命就变成越来越重要。序列比对的理论基础就是进化学说。如果两个序列之间相似性较高,就可以推测二者在进化上可能有共同的祖先,经过序列内残基的替换、残基或序列片段的确是、以及序列重组等遗传变异过程分别演化而来。

44
4.1 核酸序列的基本分析 分子量、碱基组成、限制性内切酶酶切分析、测序分析、序列转换(如将DNA序列翻译成蛋白质序列,或转换成反向互补序列等)、ORF、等电点等,均可用各种常规的核酸分析软件加以分析,如DNAStar、DNAMAN、BioEdit等。 这些核酸分析软件通常也可以用于蛋白质序列的基本分析。

45
研究项目 工具 URL 相似性搜索 Blast在线程序 重叠群组装 CAP3在线程序等 ORF预测 ORF finder在线程序等 核酸及蛋白质组成分析 DNAStar、BioEdit等软件 多序列比对 ClustalX等 进化树的建立 Mega、phylip等 多态性分析 DNAsp等

46
以DNAStar的EditSeq软件为例。其他功能大家可自学。
序列转换 DNA碱基组成 ORF查找 以DNAStar的EditSeq软件为例。其他功能大家可自学。

47
4.2 克隆测序分析 克隆测序分析是分子生物学实验日常操作之一,一般情况下单次测序将产生 bp的序列,或 bp的序列。将测序峰图识别为序列的过程称为碱基读出(base calling)。送交专业公司进行测序的结果返回后需要对所测序列进行一系列后续分析,如测序峰图的查看和载体序列的去除及序列装配等过程。当然,服务较好的测序公司后续工作做的也较好。
。送交专业公司进行测序的结果返回后需要对所测序列进行一系列后续分析,如测序峰图的查看和载体序列的去除及序列装配等过程。当然,服务较好的测序公司后续工作做的也较好。")
48
测序峰图查看 为了核实测序的准确性,往往需要对测序峰文件进行直接分析。Windows环境下最简单的峰图查看程序是澳大利亚的Chromas.exe程序,这是一个专业程序,运行快、操作简单。 其它的软件还有BioEdit和DNAMAN等也都具有该功能。 有免费的lite和注册版两种版本

49
Chromas软件查看测序峰图 打开.ab1文件。 为什么

50
开始一段序列的信号很杂乱,几乎难以辨别,主要是因为残存的染料单体造成的干扰峰所致。该干扰峰和正常序列峰重叠在一起;另外,测序电泳开始阶段电压有一个稳定期,所以经常有20-50 bp 的紧接着引物的片段读不清楚,有时甚至更长。

51
DNAMAN软件也可用来查看测序峰图

52
调节按钮 导出序列

53
4.3 核酸序列的比对分析 序列比对(Sequence alignment)是序列相似性分析的常用方法,又称序列联配。
通过将两个或多个核酸序列或蛋白序列进行比对,显示其中相似的结构域,这是进一步相似性分析的基础。通过比较未知序列与已知序列的一致性或相似性,可以预测未知序列功能。 可以用在线程序Blast进行比对,也可以用本地软件进行比对。 相似性比较:就是将待研究序列与DNA或蛋白质序列库进行比较,用于确定该序列的生物属性,也就是找出与此序列相似的已知序列是什么。完成这一工作只需要使用两两序列比较算法。常用的程序包有BLAST、FASTA等。 序列比对(Sequence alignment)是序列相似性分析的常用方法,又称序列联配。通过将两个或多个核酸序列或蛋白序列进行比对,显示其中相似的结构域,这是进一步相似性分析的基础。通过比较未知序列与已知序列的一致性或相似性,可以预测未知序列功能。
是序列相似性分析的常用方法,又称序列联配。通过将两个或多个核酸序列或蛋白序列进行比对,显示其中相似的结构域,这是进一步相似性分析的基础。通过比较未知序列与已知序列的一致性或相似性,可以预测未知序列功能。")
54
序列比对的数学模型 序列比对的实现须依赖于某个数学模型。数学模型大体可以分 为两类,一类从全长序列出发,考虑序列的整体相似性,即整 体比对;第二类考虑序列部分区域的相似性,即局部比对。 对序列从头到尾进行比较,试图使尽可能多的字符在同一列中匹配。 适用于相似度较高且长度相近的序列 如:Needleman-Wunsch算法 全局比对 不同的模型,可以从不同角度反映序列的特性,如结构、功能、进化关系等。很难断定,一个模型一定比另一个模型好,也不能说某个比对结果一定正确或一定错误,而只能说它们从某个角度反映了序列的生物学特性。此外,模型参数的不同,也可能导致比对结果的不同。 寻找序列中相似度最高的区域,也就是匹配密 度最高的部分。 适用于在某些部分相似度较高,而其他部位差 异较大的序列。速度快。 如:Smith-Waterman算法 局部比对

55
局部相似性比对 局部相似性比对的生物学基础是蛋白质功能位点往往是由较短的序列片段组成的,这些部位的序列具有相当大的保守性,尽管在序列的其它部位可能有插入、删除或突变。此时,局部相似性比对往往比整体比对具有更高的灵敏度,其结果更具生物学意义。 BLAST和FastA等常用的数据库搜索程序均采用局部相似性比对的方法,具有较快的运行速度,而基于整体相似性比对的数据库搜索程序则需要超级计算机或专用计算机才能实现。 区分这两类相似性和这两种不同的比对方法,对于正确选择比对方法是十分重要的。应该指出,在实际应用中,用整体比对方法企图找出只有局部相似性的两个序列之间的关系,显然是徒劳的;而用局部比对得到的结果也不能说明这两个序列的三维结构或折叠方式一定相同。

56
两条序列比对(pairwise alignment)
Pairwise alignment: The process of lining up two sequences to achieve maximal levels of identity(一致性) (and conservation, in the case of amino acid sequences) for the purpose of assessing the degree of similarity and the possibility of homology. 双序列比对就是排列两条序列以达到最大程度相同的过程(在氨基酸比对的情况下是最大程度的保守性)。 双序列比对的目的:衡量两个分子的相似性和同源的可能性,以便进一步揭示生物序列的功能、结构和进化的信息。 序列比对就是通过比较生物分子序列,发现它们的相似性,找出序列之间共同的区域,同时辨别序列之间的差异,从而揭示生物序列的功能、结构和进化的信息。它是科学研究中最常见最基础最经典的研究手段。
 (and conservation, in the case of amino acid sequences) for the purpose of assessing the degree of similarity and the possibility of homology. 双序列比对就是排列两条序列以达到最大程度相同的过程(在氨基酸比对的情况下是最大程度的保守性)。 双序列比对的目的:衡量两个分子的相似性和同源的可能性,以便进一步揭示生物序列的功能、结构和进化的信息。 序列比对就是通过比较生物分子序列,发现它们的相似性,找出序列之间共同的区域,同时辨别序列之间的差异,从而揭示生物序列的功能、结构和进化的信息。它是科学研究中最常见最基础最经典的研究手段。")
57
多重序列比对(multiple alignment)
多个序列的比对。 1. 不同物种中,许多基因的功能保守,序列相似性较高,通过多条序列的比较,发现保守与变异的部分 2. 可搜索更多的同源序列 3. 构建进化的树的必须步骤 4. 比较基因组学研究 两类:全局或局部的多序列比对。

58
同源性、相似性、一致性 同源性(homology)
Homology: Similarity attributed to descent from a common ancestor. 如果两个序列有一个共同的进化祖先,那么它们是同源的。这里不存在同源性的程度问题。这两条序列之间要么是同源的,要么是不同源的。 所谓同源序列,简单地说,是指从某一共同祖先经趋异进化而形成的不同序列。 同源蛋白质的氨基酸序列在三维结构上通常具有明显的相似性。 数据库搜索的基础是序列的相似性比对,而寻找同源序列则是数据库搜索的主要目的之一。 同源蛋白在三维结构上总是会有显著的相似性(similarity)。
。")
59
相似性(similarity) Similarity: The extent to which nucleotide or protein sequences are related. It is based upon identity plus conservation. 相似性是指序列比对过程中用来描述检测序列和目标序列之间相同DNA碱基或氨基酸残基顺序所占比例的高低。 相似性本身的含义,并不要求与进化起源是否同一,与亲缘关系的远近、甚至于结构与功能有什么联系。 当相似程度高于50%时,比较容易推测检测序列和目标序列可能是同源序列;而当相似性程度低于20%时,就难以确定或者根本无法确定其是否具有同源性。 而相似性(similarity)和同源性(homology)是两个完全不同的概念。不能把相似性和同源性混为一谈。所谓“具有50%同源性”,或“这些序列高度同源”等说法,都是不确切的,应该避免使用。
和同源性(homology)是两个完全不同的概念。不能把相似性和同源性混为一谈。所谓 具有50%同源性 ,或 这些序列高度同源 等说法,都是不确切的,应该避免使用。")
60
一致性(identity) Identity: The extent to which two (nucleotide or amino acid) sequences are invariant. 当两条序列同源时,它们的氨基酸序列或核苷酸序列通常有显著的一致性(identity)。 一致性反映的是两个氨基酸序列(或核苷酸序列)之间相同的程度。 因此,同源性是序列同源或不同源的一种论断(定性),而一致性和相似性是一种描述序列相关性的量(定量)。
。 一致性反映的是两个氨基酸序列(或核苷酸序列)之间相同的程度。 因此,同源性是序列同源或不同源的一种论断(定性),而一致性和相似性是一种描述序列相关性的量(定量)。")
61
Pairwise alignment of retinol-binding protein and b-lactoglobulin
1 MKWVWALLLLAAWAAAERDCRVSSFRVKENFDKARFSGTWYAMAKKDPEG 50 RBP . ||| | | | : .||||.:| : 1 ...MKCLLLALALTCGAQALIVT..QTMKGLDIQKVAGTWYSLAMAASD. 44 lactoglobulin 51 LFLQDNIVAEFSVDETGQMSATAKGRVR.LLNNWD..VCADMVGTFTDTE 97 RBP : | | | | :: | .| . || |: || |. 45 ISLLDAQSAPLRV.YVEELKPTPEGDLEILLQKWENGECAQKKIIAEKTK 93 lactoglobulin 98 DPAKFKMKYWGVASFLQKGNDDHWIVDTDYDTYAV QYSC 136 RBP || || | :.|||| | | 94 IPAVFKIDALNENKVL VLDTDYKKYLLFCMENSAEPEQSLAC 135 lactoglobulin 137 RLLNLDGTCADSYSFVFSRDPNGLPPEAQKIVRQRQ.EELCLARQYRLIV 185 RBP . | | | : || | || | 136 QCLVRTPEVDDEALEKFDKALKALPMHIRLSFNPTQLEEQCHI lactoglobulin Identity (bar) 竖线:表示相同的氨基酸残基 retinol-binding protein:视黄醇(维生素A)结合蛋白 β-lactoglobulin: β-乳球蛋白
 竖线:表示相同的氨基酸残基. retinol-binding protein:视黄醇(维生素A)结合蛋白. β-lactoglobulin: β-乳球蛋白.")
62
Pairwise alignment of retinol-binding protein and b-lactoglobulin
1 MKWVWALLLLAAWAAAERDCRVSSFRVKENFDKARFSGTWYAMAKKDPEG 50 RBP . ||| | | | : .||||.:| : 1 ...MKCLLLALALTCGAQALIVT..QTMKGLDIQKVAGTWYSLAMAASD. 44 lactoglobulin 51 LFLQDNIVAEFSVDETGQMSATAKGRVR.LLNNWD..VCADMVGTFTDTE 97 RBP : | | | | :: | .| . || |: || |. 45 ISLLDAQSAPLRV.YVEELKPTPEGDLEILLQKWENGECAQKKIIAEKTK 93 lactoglobulin 98 DPAKFKMKYWGVASFLQKGNDDHWIVDTDYDTYAV QYSC 136 RBP || || | :.|||| | | 94 IPAVFKIDALNENKVL VLDTDYKKYLLFCMENSAEPEQSLAC 135 lactoglobulin 137 RLLNLDGTCADSYSFVFSRDPNGLPPEAQKIVRQRQ.EELCLARQYRLIV 185 RBP . | | | : || | || | 136 QCLVRTPEVDDEALEKFDKALKALPMHIRLSFNPTQLEEQCHI lactoglobulin Somewhat similar (one dot) Very (two dots) 碱性氨基酸(K,R,H) 酸性氨基酸(D,E) 羟基化氨基酸(S,T) 疏水性氨基酸(W,F,Y,L,I,V,M,A) 两点:表示性质非常相似的氨基酸残基 单点:相似程度较少 第一行的比对中,11个相同,3个相似,因此两条序列的Identity=11/44=25%,Similarrity=14/44=32%
 Very. (two dots) 碱性氨基酸(K,R,H) 酸性氨基酸(D,E) 羟基化氨基酸(S,T) 疏水性氨基酸(W,F,Y,L,I,V,M,A) 两点:表示性质非常相似的氨基酸残基. 单点:相似程度较少. 第一行的比对中,11个相同,3个相似,因此两条序列的Identity=11/44=25%,Similarrity=14/44=32%")
63
间隙(Gaps) Positions at which a letter is paired with a null are called gaps. • Gap scores are typically negative. 应用上:间隙的加入是为了使两个序列在总体长度上相等,使得两个蛋白质能进行全长比对; 生物学上:间隙的加入能反映进化上发生的变化,如:删除和插入突变; 通过视觉检查来比对两条序列是十分困难的。 In BLAST, it is rarely necessary to change gap values from the default. 63

64
Pairwise alignment of retinol-binding protein and b-lactoglobulin
1 MKWVWALLLLAAWAAAERDCRVSSFRVKENFDKARFSGTWYAMAKKDPEG 50 RBP . ||| | | | : .||||.:| : 1 ...MKCLLLALALTCGAQALIVT..QTMKGLDIQKVAGTWYSLAMAASD. 44 lactoglobulin 51 LFLQDNIVAEFSVDETGQMSATAKGRVR.LLNNWD..VCADMVGTFTDTE 97 RBP : | | | | :: | .| . || |: || |. 45 ISLLDAQSAPLRV.YVEELKPTPEGDLEILLQKWENGECAQKKIIAEKTK 93 lactoglobulin 98 DPAKFKMKYWGVASFLQKGNDDHWIVDTDYDTYAV QYSC 136 RBP || || | :.|||| | | 94 IPAVFKIDALNENKVL VLDTDYKKYLLFCMENSAEPEQSLAC 135 lactoglobulin 137 RLLNLDGTCADSYSFVFSRDPNGLPPEAQKIVRQRQ.EELCLARQYRLIV 185 RBP . | | | : || | || | 136 QCLVRTPEVDDEALEKFDKALKALPMHIRLSFNPTQLEEQCHI lactoglobulin Internal gap Terminal gap 共八处间隙。 64

65
4.3.1用Blast在线程序进行序列比对 BLAST是一个序列数据库搜索程序家族,BLAST检索的网络资源较多:
在NCBI上作blast存在着速度和安全的问题,因此也可以将数据库下载到本地进行本地Blast。 NCBI的下载网址:ftp://ftp.ncbi.nih.gov/blast/ Blast介绍文档

66
Blast 序列两两比对分析是最简单、最基本的对齐分析。
Blast(基本局域联配搜索工具, Basic Local Alignment Search Tool)是一个局部比对搜索工具,用来确定一条查询序列和一个数据库的比对,最早的版本不引入间隙,但现在所用的版本已经允许比对中引入间隙。 “Bl2Seq”是NCBI上Blast程序的一部分,允许两条序列之行局部双序列比对,使用这个程序执行蛋白质(或DNA序列)的双序列比对非常容易。
是一个局部比对搜索工具,用来确定一条查询序列和一个数据库的比对,最早的版本不引入间隙,但现在所用的版本已经允许比对中引入间隙。 Bl2Seq 是NCBI上Blast程序的一部分,允许两条序列之行局部双序列比对,使用这个程序执行蛋白质(或DNA序列)的双序列比对非常容易。")
67
NCBI中BLAST检索服务 BLASTN:核酸比对 BLASTP:蛋白质比对 BLASTX:用核酸搜索蛋白质数据库
TBLASTX:核酸比对 Blast在线程序的网址

68
也可用BLAST工具进行两条序列之间的比对
网址:

70
竖线:一致性(identities) 空白:错配
横线:间隙(gap),插入或删除突变。
,插入或删除突变。")
71
双序列比对的显著性:一致性百分比 核酸和蛋白质序列进行对库检索的结果中是否具有生物学意义是一个很重要的问题。蛋白质序列对齐分析得到的结论是:
对于有70个氨基酸残基的比对,40%的氨基酸一致性(identities)是一个认为两个蛋白同源的合理阈值,即它们一般具有相类似的生物学性质;在此标准之下,两条蛋白质序列可能具有相似的功能,也可能是性质上完全不同的蛋白质。 对于DNA序列需要具有75%以上的一致性才可能具有潜在的生物学意义。 最早认为:考虑有70个氨基酸长度的比对,如果它们共享25%的氨基酸一致性,则认为这两条序列“显著相关”,这种做法比较普遍。但后来有人认为(Brenner等,1998)这可能会产生错误,部分是因为当今的分子序列数据库变得越来越大,使得随机找到这样比对的可能性增大。因此,对于有70个氨基酸残基的比对,40%氨基酸一致性是一个认为两个蛋白同源的合理阈值。如果两个蛋白质比对后,只有10~20%的一致性,那么可以认为它们是完全不相关的。
是一个认为两个蛋白同源的合理阈值,即它们一般具有相类似的生物学性质;在此标准之下,两条蛋白质序列可能具有相似的功能,也可能是性质上完全不同的蛋白质。 对于DNA序列需要具有75%以上的一致性才可能具有潜在的生物学意义。 最早认为:考虑有70个氨基酸长度的比对,如果它们共享25%的氨基酸一致性,则认为这两条序列 显著相关 ,这种做法比较普遍。但后来有人认为(Brenner等,1998)这可能会产生错误,部分是因为当今的分子序列数据库变得越来越大,使得随机找到这样比对的可能性增大。因此,对于有70个氨基酸残基的比对,40%氨基酸一致性是一个认为两个蛋白同源的合理阈值。如果两个蛋白质比对后,只有10~20%的一致性,那么可以认为它们是完全不相关的。")
72
黏贴我们刚才下载的“neuraminidase”序列
也可用BLAST工具进行多重序列比对 蛋白质序列比对工具blastp 黏贴我们刚才下载的“neuraminidase”序列 数据库可选,然后点击“blast”

73
提示了这个蛋白的保守结构,及相应的蛋白质家族
与检索序列一致性最高的前100个序列。一致性的高低反映到颜色上就是: 红色>紫色>绿色>蓝色>黑色

74
结果显示:比对出来的都是乙型流感病毒的“neuraminidase”序列,且一致性最高的序列排最前面,可从中挑选合适的序列下载,用于后续的进化分析等。

75
这里一排的数值显示这条序列与目的序列之间的比较关系。

76
4.3.2 也可用本地化软件进行序列比对 常用clustalX软件做多重比对分析等( clustalw 则是在线软件,网址: Clustal是一个单机版的基于渐进比对的多序列比对工具。其基本思想就是基于相似序列通常具有进化相关性的这一假设。 当然,DNAStar、DNAMan等软件也可以进行比对。 在比对过程中,先对所有的序列进行两两比对并计算它们相似性分值,然后根据相似性分值将它们分成若干组,并在每组之间进行比对,计算相似性分值。根据相似性分值继续分组比对,直到得到最终比对结果。在比对过程中,相似性程度较高的序列先进行比对而距离较远的序列添加在后面。 生物软件网:

77
快速的序列两两比对,计算序列间的距离,获得一个距离矩阵。
ClustalX软件 Clustal软件工作原理 CLUSTAL是一种渐进的比对方法,先将多个序列两两比对构建距离矩阵,反映序列之间两两关系;然后根据距离矩阵计算产生系统进化指导树,对关系密切的序列进行加权;然后从最紧密的两条序列开始,逐步引入临近的序列并不断重新构建比对,直到所有序列都被加入为止。 在Clustal软件中输入多个序列 快速的序列两两比对,计算序列间的距离,获得一个距离矩阵。 在比对过程中,先对所有的序列进行两两比对并计算它们相似性分值,然后根据相似性分值将它们分成若干组,并在每组之间进行比对,计算相似性分值。根据相似性分值继续分组比对,直到得到最终比对结果。在比对过程中,相似性程度较高的序列先进行比对而距离较远的序列添加在后面。 采用邻接法(NJ)构建一个树(引导树) 根据引导树,渐进比对多个序列。
构建一个树(引导树) 根据引导树,渐进比对多个序列。")
78
Clustal的输入输出格式 输入序列的格式比较灵活,可以是前面介绍过的FASTA格式,还可以是PIR、SWISS-PROT、GDE、Clustal、GCG/MSF、RSF等格式。 输出格式也可以选择,有ALN、GCG、PHYLIP和NEXUS等,用户可以根据自己的需要选择合适的输出格式。 2.两种工作模式。 a.多序列比对模式。 b.剖面(profile)比对模式。
比对模式。")
79
用clastalX软件进行两条序列之间的比对
载入的序列常见的是fasta格式,存储在记事本(.txt)中,也可以是直接以.fasta格式存储的文件。
中,也可以是直接以.fasta格式存储的文件。")
80
参数可以选择,或者默认。

82
Clustal比对后的结果

83
用ClustalX进行多重序列的比对,步骤相同
以多个物种的抗坏血酸过氧化物酶的的蛋白质序列进行比对为例 如果要在DNA序列比对和对应编码的蛋白质序列比对之间选择,通常是比较蛋白质序列更有用。 因为DNA序列的许多改变不会改变对应的氨基酸;而且许多氨基酸有相似的生物物理性质。 将要比对的多个序列以Fasta格式存储于同一个文本中。

84
载入多个序列后,选择输出选项,选择输出格式。或者在比对完成之后,在“文件”中选择“序列另存为”,同样可以选择合适的输出格式。

85
选择“进行完全比对”,输出的文件路径自动与原始的序列文件的路径一致。

86
Clustal比对结果 星号:完全一致 两点:性质很相似 一点:性质较相似 峰:表示一致程度高 谷:表示一致程度低

87
4.3.3 Clustal比对结果的编辑 Clustal比对之后的结果,可用于分子进化分析使用。
也可以采用其他软件进行编辑,如BioEdit软件、GeneDoc软件,但必须注意所用软件的输入文件的格式。 BioEdit软件不能识别“.aln”格式,但可识别“.pir”或“.phy”格式文件。 也可以采用一些在线的着色软件来编辑Clustal比对结果。如Boxshade软件,网址:

88
本地软件编辑比对结果:以BioEdit软件为例

91
相当于“复制”键,可黏贴到其他文件,如“word、PowerPoint”等
各种调整图形的参数可选。

92
这是黏贴后的结果 当然,还有其他软件可用于比对结果的编辑,大家可去自行搜索一下。

93
BioEdit也可以用于核酸序列的常规分析
这里是与核酸有关的各种分析,如碱基组成、ORF、序列转换、内切酶位点等

94
4.3.4 电子克隆与EST序列 电子克隆:以物种A的某蛋白质序列X作为种子序列,对物种B的EST数据库BLAST比对,所得的所有EST序列用程序拼接(CAP3、DNAMAN、DNAStar等)成一条序列后,再以新拼得的序列为种子重复BLAST物种B的EST数据库,将新的EST数据与原来拼好的序列再次拼接,重复这种BLAST、拼接的过程,直至没有新的EST序列产生。最终所拼得的序列拿去分析其ORF、起始密码子等,通常能得到物种B的X基因。
成一条序列后,再以新拼得的序列为种子重复BLAST物种B的EST数据库,将新的EST数据与原来拼好的序列再次拼接,重复这种BLAST、拼接的过程,直至没有新的EST序列产生。最终所拼得的序列拿去分析其ORF、起始密码子等,通常能得到物种B的X基因。")
95
4.3.5引物设计 Primer Premier (引物设计) Oligo 6 (引物评价) Dnastar(引物设计)
也可用NCBI上的primer-blast在线程序

96
引物设计的原则 长度:15-30bp,常用的是18-24bp 引物与模板的序列要紧密互补 引物自身不要形成二级结构 引物之间避免形成稳定的二聚体 引物5’端可添加内切酶位点及其他碱基修饰 引物3’端最后一个碱基不要用“A”,也不要连续的G或C 引物不能在模板的非目的位点引发DNA聚合反应(即错配。因此目的序列最好不要有与引物3’端相似性较高的序列) GC含量:一般为40-60% ∆G 值:指DNA 双链形成所需的自由能,该值反映了双链结构内部碱基对的相对稳定性。应当选用3’端∆G 值较低(绝对值不超过9),而5’端和中间∆G 值相对较高的引物。引物的3’端的∆G 值过高(说明容易结合),容易在错配位点形成双链结构并引发DNA 聚合反应。 ∆G 值越高越容易结合
 GC含量:一般为40-60% ∆G 值:指DNA 双链形成所需的自由能,该值反映了双链结构内部碱基对的相对稳定性。应当选用3’端∆G 值较低(绝对值不超过9),而5’端和中间∆G 值相对较高的引物。引物的3’端的∆G 值过高(说明容易结合),容易在错配位点形成双链结构并引发DNA 聚合反应。 ∆G 值越高越容易结合.")
97
功能区:包含查找引物、DNA结构motif、酶切位点等
Primer Premier 功能区:包含查找引物、DNA结构motif、酶切位点等 激活序列,可翻译成蛋白质 输入的序列区域

98
引物参数的选择 点击“功能区”中的“primer”后,在出现的框中点击“search”,然后在新出现的参数框中选择合适的引物参数,确定。

99
这是所选择的第2对引物的结果评价,包括引物长度、所处位置、Tm值、是否会形成茎环结构、是否有引物二聚体、是否会错误引发等
显示引物对的结果 可对引物进行编辑 这是所选择的第2对引物的结果评价,包括引物长度、所处位置、Tm值、是否会形成茎环结构、是否有引物二聚体、是否会错误引发等

100
若引物设计无误,可打开菜单栏的“function”中“save to database”,将引物存于引物库中。

101
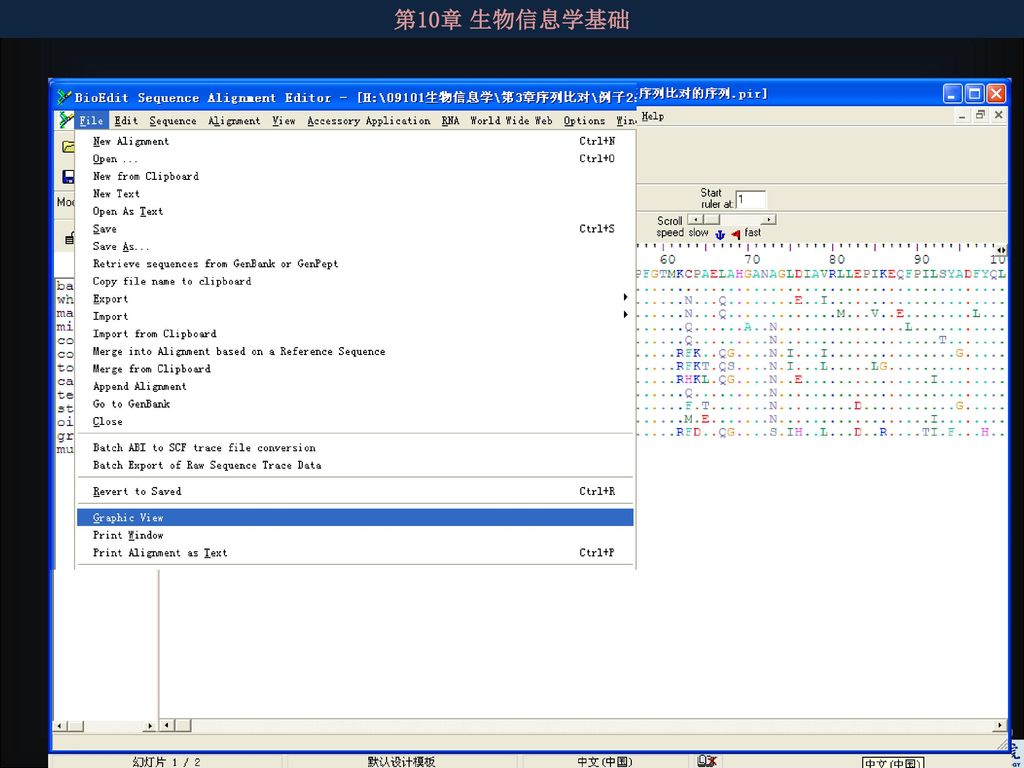
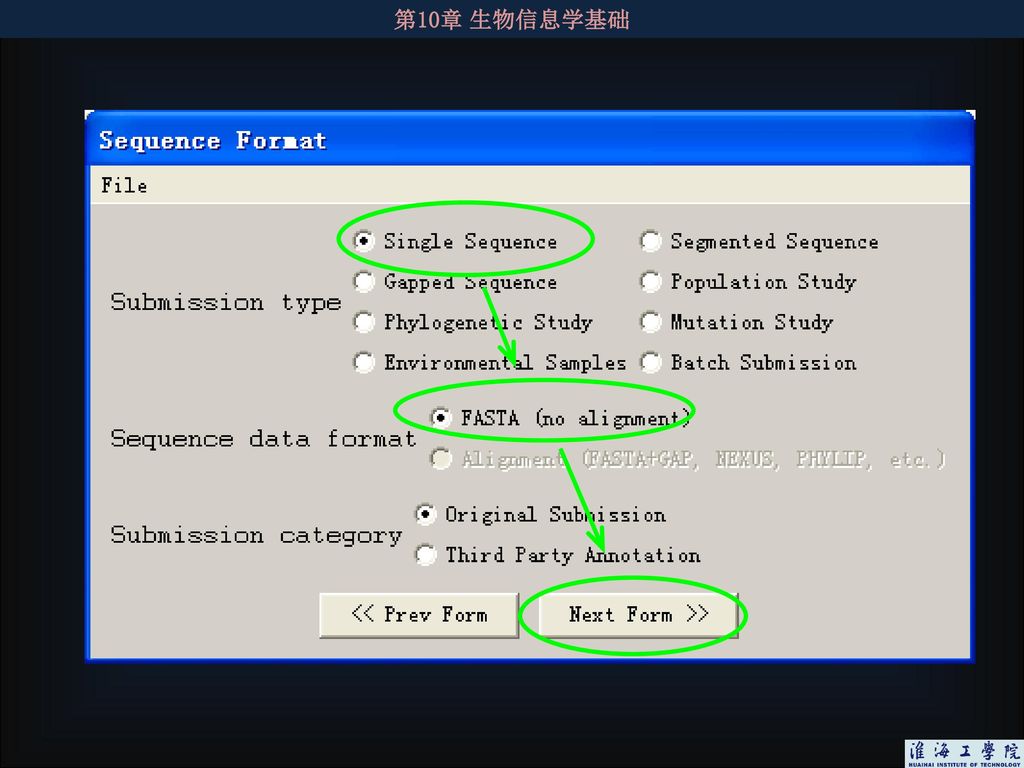
4.3.6 如何向数据库提交核酸序列 几乎所有杂志都要求提供目标序列的提交信息,因此获得的序列一定要向核酸数据库提交。序列提交方法相对较简单,各大数据库都有详细提交说明,根据说明step-by-step地做即可完成提交。提交序列后仍可对提交的序列进行修改,如果所提交的序列不合乎要求将会得到工作人员的通知。 序列被接受后将被赋予一个序列接受号,用于在出版论文中引用。 向NCBI提交序列有两个常用的软件:Sequin(要下载安装,提交的序列可批量且较复杂,如各种编码蛋白质的基因序列)和BankIt(在线提交程序,适合单个或少量的序列,且序列的注释不复杂,不必用到序列分析,如16S RNA序列)
和BankIt(在线提交程序,适合单个或少量的序列,且序列的注释不复杂,不必用到序列分析,如16S RNA序列)")
102
4.3.6.1 BankIt在线提交序列 在线提交页面:http://www.ncbi.nlm.nih.gov/BankIt/) 特点:
适合单个或少量几个的提交 在线的方式提交,无需下载个软件 提交的序列的注释不复杂 提交的序列不需要用到序列的分析,如:16S RNA

103
NCBI主页上通过submission

104
如何提交各种数据的使用说明

105
左侧栏是各种序列的提交范本 需要注册才能在线提交

106
BankIt在线提交页面的最下方: 填入待提交序列的大小,然后点击:NEW

107
信息填写的主页 输入序列长度并点击New之后,会生成一个bankit号。下面根据需要填写4个必需的内容。包括提交者的必要信息、序列参考信息、来源信息、具体序列。

108
…………

109
以上内容填写无误后,点击“save this form”保存于本地电脑。
然后再点击“validate and continue”

110
如果填的资料有误,或漏填了,会有ERROR的提醒。像名字,电话,Email,序列等都是必须的。
最后NCBI会给您发个邮件,里面是类似于GenBank格式的序列(其实是提交的序列内容,但是BankIt号),等过2、3个星期后如果没有问题就能得到GenBank的登录号了,如果有问题NCBI会通过邮件和提交者联系。
,等过2、3个星期后如果没有问题就能得到GenBank的登录号了,如果有问题NCBI会通过邮件和提交者联系。")
111
点击,进入下载页面,根据自身电脑所装系统下载合适的软件。
Sequin软件提交序列 用Sequin的好处就是可以保存下来,随时修改。直到满意后再向NCBI提交。 点击,进入下载页面,根据自身电脑所装系统下载合适的软件。

112
双击释放程序 双击打开程序 帮助文件

113
首页简介 选择数据库 版本信息 提交新序列 打开已有提交文件 帮助 退出

114
提交新序列 论文题目,实际上是序列的题目

115
通讯作者及联系方式

116
作者列表及排序 社团或协会等

117
作者所属机构及城市、省份、国家等

119
格式要求

120
对输入内容修改补充

121
格式要求 输入错误时会提醒修改

122
不填也可!

123
自己检测 通过双击修改

124
软件检测修改

125
发现错误 通过双击修改

126
准备提交

127
接下来的工作就是发邮件,以附件的形式将欲提交的序列提交上去。难度稍微大一点的就是正确无歧义地与数据库工作人员交流!
后续工作是对自己提交的序列进行更正和更新。更新的方法也比较简单,可打开自己已有的提交文件进行编辑,也可以直接从数据库中下载已经提交的序列进行编辑。

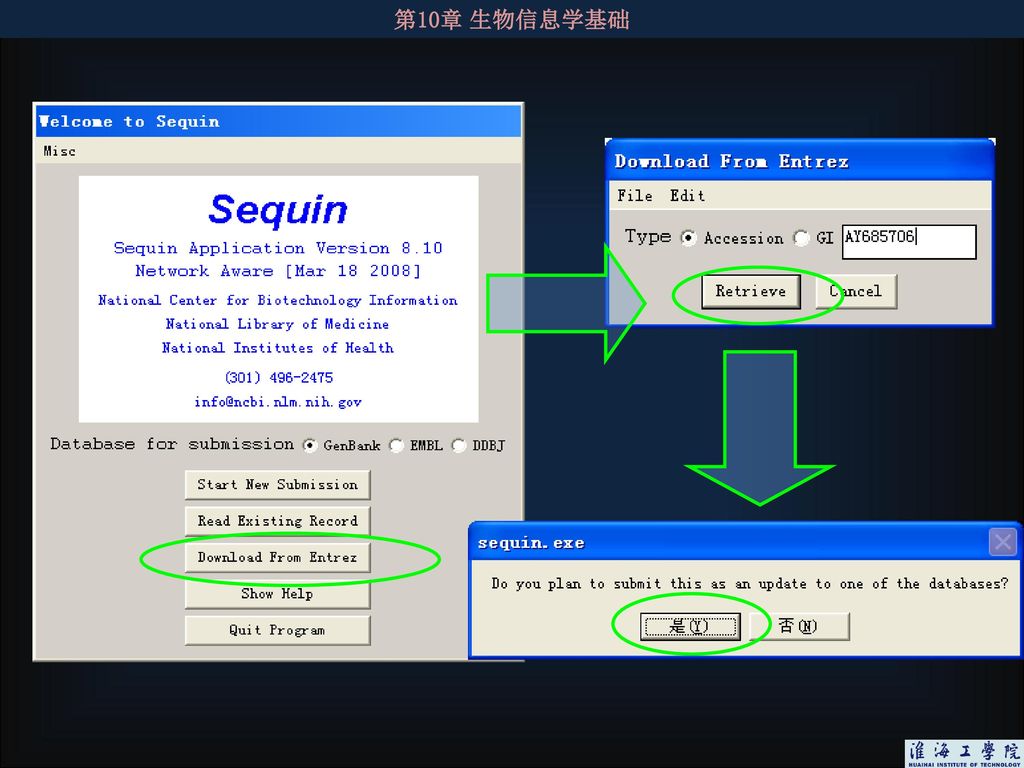
128
利用Sequin从数据库下载已有序列进行更正或更新!

130
填写信息!

132
5 蛋白质数据分析

133
研究项目 工具 URL 核酸及蛋白质组成分析 DNAStar、BioEdit等 多序列比对 ClustalX软件等 进化树的建立 Mega软件等 信号肽预测 SignalP4.1软件 疏水性分析 ProtScale程序 保守结构域分析 prosite程序 二级结构预测 SOPMA 三级结构预测 Geno3D程序、Swiss-Model等 通常在线提交序列,设置一定参数后,就能获得结果,有时还需填入邮箱。

134
6 分子进化分析

135
6.1 一些概念 系统发育(种系发生、系统发生):指生物形成或进化的历史;
系统发育学:研究物种之间的进化关系,基本思想是比较物种的特征,并认为特征相似的物种在遗传学上接近。研究结果往往以系统发生树(系统发育树)表示,用它描述物种之间的进化关系。 通过对生物学数据的建模提取特征,进而比较这些特征,研究生物形成或进化的历史。在分子水平上进行系统发生分析具有许多优势,所得到的结果更加科学、可靠。
表示,用它描述物种之间的进化关系。 通过对生物学数据的建模提取特征,进而比较这些特征,研究生物形成或进化的历史。在分子水平上进行系统发生分析具有许多优势,所得到的结果更加科学、可靠。")
136
经典系统发育学研究所涉及的特征主要是生物的表型特征(指形态学的或结构的特征)。通过表型比较来推断生物体的基因型,研究物种之间的进化关系。
系统发育分析早在达尔文时代就已经开始 经典系统发育学研究所涉及的特征主要是生物的表型特征(指形态学的或结构的特征)。通过表型比较来推断生物体的基因型,研究物种之间的进化关系。 利用表型特征的局限性:表型相似并不总是反映基因相似;对于许多生物体很难检测到可用来进行比较的表型特征;如何选择表性特征。 有关生物群体伴随时间的推移不断改变,从而导致其后代在结构和功能上不同于祖先的理论称为进化理论。 1859年查尔斯.达尔文发表了其标志性的学术著作——《在自然选择意义下的物种起源》,亦可称为《在生存竞争中保留优势物种》。 进化可以被定义为生物体系统发生的过程。
。通过表型比较来推断生物体的基因型,研究物种之间的进化关系。 利用表型特征的局限性:表型相似并不总是反映基因相似;对于许多生物体很难检测到可用来进行比较的表型特征;如何选择表性特征。 有关生物群体伴随时间的推移不断改变,从而导致其后代在结构和功能上不同于祖先的理论称为进化理论。 1859年查尔斯.达尔文发表了其标志性的学术著作——《在自然选择意义下的物种起源》,亦可称为《在生存竞争中保留优势物种》。 进化可以被定义为生物体系统发生的过程。")
137
分子系统发育分析 蛋白质序列和DNA序列的测序为分子系统发生分析提供了可靠的数据。
分子进化:是生物分子层次上的进化,分子系统学是利用生物大分子(蛋白质、核酸)的信息推断生物进化历史,或者说重建系统发育关系,并以系统树形式表示出来。 蛋白质序列和DNA序列的测序为分子系统发生分析提供了可靠的数据。 基本原理:从一条序列转变为另一条序列所需要的变换越多,那么,这两条序列的相关性就越小,从共同祖先分歧的时间就越早,进化距离就越大;相反,两个序列越相似,那么它们之间的进化距离就可能越小。为了便于分析,一般假设序列变化的速率相对恒定。 在分子水平上研究生物之间的关系早在20世纪初就开始了。 对于给定的分类单元数,有很多棵可能的系统发生树,但是只有一棵树是正确的。系统发生分析的目标,就是——寻找这棵正确的树。
的信息推断生物进化历史,或者说重建系统发育关系,并以系统树形式表示出来。 蛋白质序列和DNA序列的测序为分子系统发生分析提供了可靠的数据。 基本原理:从一条序列转变为另一条序列所需要的变换越多,那么,这两条序列的相关性就越小,从共同祖先分歧的时间就越早,进化距离就越大;相反,两个序列越相似,那么它们之间的进化距离就可能越小。为了便于分析,一般假设序列变化的速率相对恒定。 在分子水平上研究生物之间的关系早在20世纪初就开始了。 对于给定的分类单元数,有很多棵可能的系统发生树,但是只有一棵树是正确的。系统发生分析的目标,就是——寻找这棵正确的树。")
138
梦想走进现实:How? 1. 最理想的方法:化石!—— 零散、不完整
2. 比较形态学和比较生理学:确定大致的进化框架 —— 但细节存很多的争议(属于经典系统发生学) 传统上,构建系统树是利用化石证据,通过严密的推理或计算,来表示不同物种间遗传距离及进化顺序。但是由于化石的零散性和不完整性,使大多数研究者转向比较形态学和比较生理学的方法。通过这两种方法,经典进化学家己得出有机体进化历史的主要框架。然而形态和生理性状是如此复杂,以致不可能产生一幅进化历史的清晰图像。
 传统上,构建系统树是利用化石证据,通过严密的推理或计算,来表示不同物种间遗传距离及进化顺序。但是由于化石的零散性和不完整性,使大多数研究者转向比较形态学和比较生理学的方法。通过这两种方法,经典进化学家己得出有机体进化历史的主要框架。然而形态和生理性状是如此复杂,以致不可能产生一幅进化历史的清晰图像。")
139
第三种方案:分子进化 Linus Pauling 1. 1964年,Linus Pauling提出分子进化理论
2. DNA & RNA: 4种碱基;蛋白质分子:20种氨基酸 3. 发生在分子层面的进化过程:DNA, RNA和蛋白质分子 4. 基本假设:核苷酸和氨基酸序列中含有生物进化历史的全部信息 分子生物学的进展大大地改变了这种局面。由于所有生物的蓝图都用DNA(在某些病毒中用RNA)来书写,因而人们得以通过比较DNA来研究它们的进化关系,即DNA分子进化。 分子进化从组织层次上说是生物组织的基础层次的进化,有两个显著特点:进化速率相对恒定和进化的保守性。生物大分子进化速率相对恒定是建立分子系统树的理论前提。近年来一些重要的分子系统学研究进展主要有:(1)生物进化系统的3条主线,即30多亿年以前,整个生物界就分为原始真核细胞生物、原始真细菌和原始古细菌三大界。(2)分子系统学的研究支持了真核细胞的共生起源说。(3)分子系统学的研究结果大部分与传统的根据表型比较而建立的系统树相符合,但也有一些与传统的系统进化观点不吻合的结论。(4)揭示了一些过去表型系统树上模糊不明的或争论未决的系统进化问题。 1954年诺贝尔化学奖得主Linus Pauling在1960年代初开创性地展开的基于直系同源蛋白序列比对的分子进化与分子钟研究。通过直系同源蛋白质之间比较来确定物种之间的亲缘关系。
来书写,因而人们得以通过比较DNA来研究它们的进化关系,即DNA分子进化。 分子进化从组织层次上说是生物组织的基础层次的进化,有两个显著特点:进化速率相对恒定和进化的保守性。生物大分子进化速率相对恒定是建立分子系统树的理论前提。近年来一些重要的分子系统学研究进展主要有:(1)生物进化系统的3条主线,即30多亿年以前,整个生物界就分为原始真核细胞生物、原始真细菌和原始古细菌三大界。(2)分子系统学的研究支持了真核细胞的共生起源说。(3)分子系统学的研究结果大部分与传统的根据表型比较而建立的系统树相符合,但也有一些与传统的系统进化观点不吻合的结论。(4)揭示了一些过去表型系统树上模糊不明的或争论未决的系统进化问题。 1954年诺贝尔化学奖得主Linus Pauling在1960年代初开创性地展开的基于直系同源蛋白序列比对的分子进化与分子钟研究。通过直系同源蛋白质之间比较来确定物种之间的亲缘关系。")
140
分子进化的模式 1. DNA突变的模式:替代,插入,缺失,倒位
2. 核苷酸替代:转换 (Transition) & 颠换 (Transversion) 3. 基因复制:多基因家族的产生以及伪基因的产生 A. 单个基因复制 – 重组或者逆转录 B. 染色体片断复制 C. 基因组复制 在分子水平上,进化是一种伴随着突变的自然选择过程。分子进化理论着重于研究不同系统发育树分子上基因和蛋白质的变化方式。
 & 颠换 (Transversion) 3. 基因复制:多基因家族的产生以及伪基因的产生. A. 单个基因复制 – 重组或者逆转录. B. 染色体片断复制. C. 基因组复制. 在分子水平上,进化是一种伴随着突变的自然选择过程。分子进化理论着重于研究不同系统发育树分子上基因和蛋白质的变化方式。")
141
分子进化研究的目的 1. 从物种的一些分子特性出发,构建系统发育树,进而了解物种之间的生物系统发生的关系 —— tree of life; 物种分类 2. 大分子功能与结构的分析:同一家族的大分子,具有相似的三级结构及生化功能,通过序列同源性分析,构建系统发育树,进行相关分析;功能预测 Tree of Life: 16S rRNA 3. 进化速率分析:例如,HIV的高突变性;哪些位点易发生突变?

142
Out of Africa 人类迁移的路线 53个人的线粒体基因组(16,587bp)
")
143
6.2 分子进化树的一些概念 末端节点 分支/世系 代表最终分类,可以是物种,群体,或者蛋白质、DNA、RNA分子等 祖先节点/树根
B C D 通过比较生物大分子序列差异的数值构建的系统树称为分子系统树(molecular phylogenetic tree)。图型中。分支的末端和分支的联结点成为结(node),代表生物类群,分支末端的结代表仍生存的种类。 祖先节点/树根 E 内部节点/分歧点,该分支可能的祖先结点
。图型中。分支的末端和分支的联结点成为结(node),代表生物类群,分支末端的结代表仍生存的种类。 祖先节点/树根. E. 内部节点/分歧点,该分支可能的祖先结点.")
144
系统发育树:三种类型 枝长无意义 时间 遗传变化 以上三种类型的系统发育树表示相同的分支状况,相同的进化关系 分支树 进化树 时间度量树
分支树 进化树 时间度量树 6 Taxon B Taxon B Taxon B 1 Taxon C Taxon C Taxon C 1 Taxon A Taxon A Taxon A Taxon D Taxon D Taxon D Taxon:分类、门类;复数为taxa 枝长无意义 遗传变化 时间 以上三种类型的系统发育树表示相同的分支状况,相同的进化关系

145
系统发育树的种类:有根树、无根树 理论上,一个DNA序列在物种形成或基因复制时,分裂成两个子序列,因此系统发育树一般是二歧的。
一般考虑二歧的树结构:二歧树 拓扑结构: 有根树:反映时间顺序 无根树:反映距离 a b c d 节点: 内部节点 外部节点 a b c d 分支: 内部分支 外部分支

146
有根树:反映末端节点(物种、蛋白质、DNA等)在进化上的时间关系
有根树是具有方向的树,包含唯一的节点,将其作为树中所有物种的最近共同祖先。 有根树:反映末端节点(物种、蛋白质、DNA等)在进化上的时间关系
在进化上的时间关系.")
147
无根树:反映末端节点(物种、蛋白质、DNA)之间在进化上的距离
把有根树去掉根即成为无根树。一棵无根树在没有其他信息(外群)或假设(如假设最大枝长为根)时不能确定其树根。无根树是没有方向的,其中线段的两个演化方向都有可能。 无根树:反映末端节点(物种、蛋白质、DNA)之间在进化上的距离
或假设(如假设最大枝长为根)时不能确定其树根。无根树是没有方向的,其中线段的两个演化方向都有可能。 无根树:反映末端节点(物种、蛋白质、DNA)之间在进化上的距离.")
148
无根树,有根树,外群 根 无根树 最常用的确定树根的方法是通过一个外群来确定树根 有根树 外群(outgroup) bacteria
archaea eukaryote archaea 无根树 archaea eukaryote eukaryote 最常用的确定树根的方法是通过一个外群来确定树根 eukaryote bacteria 外群(outgroup) 当类群数较大时,选出真实树的拓扑结构十分困难。 最常用的确定树根的方法是使用一个或多个无可争议的同源物种作为[外群](英文outgroup),这个外群要足够近,以提供足够的信息,但又不能太近以致不能和树中的种类相混。 archaea archaea archaea 有根树 eukaryote eukaryote 根 eukaryote eukaryote
 当类群数较大时,选出真实树的拓扑结构十分困难。 最常用的确定树根的方法是使用一个或多个无可争议的同源物种作为[外群](英文outgroup),这个外群要足够近,以提供足够的信息,但又不能太近以致不能和树中的种类相混。 archaea. archaea. archaea. 有根树. eukaryote. eukaryote. 根. eukaryote. eukaryote.")
149
外群 (Outgroup)的选择 1. 选择一个或多个已知与分析序列关系较远的序列作为外群 2. 外群可以辅助定位树根
3. 外群序列与其他序列间的差异,必须比其他序列之间的差异更显著 bacteria 外群(outgroup) archaea eukaryote
 archaea. eukaryote.")
150
6.3 系统发育树重建分析步骤 多序列比对(clustal) 选择合适的建树方法以及替代模型 构建进化树 进化树评估(bootstrap)
自动比对,手工校正 构建系统发育树的步骤一般是:先进行序列比对;再利用比对的结构用合适的算法求出遗传距离;最后再根据遗传距离构建发育树。 构建进化树 进化树评估(bootstrap)
")
151
系统发生分析和多重序列比对 分子系统发生分析的目的是探讨物种之间的进化关系,其分析的对象往往是一组同源的序列。这些序列取自于不同生物基因组的共同位点。 序列比对是进行同源分析的一种基本手段,是进行系统发生分析的基础,一般采用基于两两比对渐进的多重序列比对方法,如ClustalW程序。通过序列的比对,可以分析序列之间的差异,计算序列之间的距离。

152
一般来讲,如果模型合适,ML的效果较好。近缘序列,可采用MP法(最大简约法),因为用的假设最少。远缘序列,一般用NJ或ML。
相似程度高?Yes。用MP法。 获得数据 多序列比对 (最大简约法) 获得多个序列 多序列比对 相似程度较高?Yes。用距离法。 一般来讲,如果模型合适,ML的效果较好。近缘序列,可采用MP法(最大简约法),因为用的假设最少。远缘序列,一般用NJ或ML。 NJ法(Neighbor-Joining Method,距离法的一种):NJ法本质上是一种寻找最优拓扑结构的谱系聚类算法。同时给出系统发育树的拓扑结构以及分支的长度。 优点: 1、可以较快地构建系统树; 2、适用于分析较大的数据集(不一定近缘,但明显具有较高的序列相似性); 3、能够较方便地进行自展(Bootstrap)检验。 (距离法) 相似程度较低。用ML法。 教材P124 (最大似然法)
 获得多个序列. 多序列比对. 相似程度较高?Yes。用距离法。 一般来讲,如果模型合适,ML的效果较好。近缘序列,可采用MP法(最大简约法),因为用的假设最少。远缘序列,一般用NJ或ML。 NJ法(Neighbor-Joining Method,距离法的一种):NJ法本质上是一种寻找最优拓扑结构的谱系聚类算法。同时给出系统发育树的拓扑结构以及分支的长度。 优点: 1、可以较快地构建系统树; 2、适用于分析较大的数据集(不一定近缘,但明显具有较高的序列相似性); 3、能够较方便地进行自展(Bootstrap)检验。 (距离法) 相似程度较低。用ML法。 教材P124. (最大似然法)")
153
MP法(最大简约法) 1)序列位点不存在回复突变、平行突变; 2)被分析的序列较长,核苷酸或氨基酸数目很大; 3)序列的相似度较高(通常是近缘序列); 4)核苷酸或氨基酸替代速率较稳定。 优点:最大简约法不需要在处理核苷酸或者氨基酸替代的时候引入假设(替代模型)。此外,最大简约法对于分析某些特殊的分子数据如插入、缺失等序列有用。 缺点:在分析的序列位点上没有回复突变或平行突变,且被检验的序列位点数很大的时候,最大简约法能够推导获得一个很好的进化树。然而在分析序列上存在较多的回复突变或平行突变,而被检验的序列位点数又比较少的时候,最大简约法可能会给出一个不合理的或者错误的进化树推导结果。 构建MP树,最好的工具是PAUP,但该程序属于商业软件,并不对学术免费。 MEGA和PHYLIP也可以用来构建进化树。
。此外,最大简约法对于分析某些特殊的分子数据如插入、缺失等序列有用。 缺点:在分析的序列位点上没有回复突变或平行突变,且被检验的序列位点数很大的时候,最大简约法能够推导获得一个很好的进化树。然而在分析序列上存在较多的回复突变或平行突变,而被检验的序列位点数又比较少的时候,最大简约法可能会给出一个不合理的或者错误的进化树推导结果。 构建MP树,最好的工具是PAUP,但该程序属于商业软件,并不对学术免费。 MEGA和PHYLIP也可以用来构建进化树。")
154
NJ法——距离法的一种 最常用的距离法——Neighbor-Joining Method (NJ法/邻接法)
在利用NJ法构建进化树时,需要选择合适的概率模型,由两条序列的差异值构建它们的进化距离。对于蛋白质序列以及DNA序列,两者模型的选择是不同的。 对于蛋白质的序列,一般选择Poisson Correction(泊松修正)这一模型。 而对于核酸序列,一般选择Kimura 2-parameter (Kimura-2参数) 模型。 构建NJ树,可以用PHYLIP(这个软件写得有点问题,运算非常慢,界面不友好,并且Bootstrap检验不方便)或者MEGA。
这一模型。 而对于核酸序列,一般选择Kimura 2-parameter (Kimura-2参数) 模型。 构建NJ树,可以用PHYLIP(这个软件写得有点问题,运算非常慢,界面不友好,并且Bootstrap检验不方便)或者MEGA。")
155
最大似然法 (ML) 最大似然法是一个比较成熟的参数估计的统计学方法,具有很好的统计学理论基础,在当样本量很大的时候,似然法可以获得参数统计的最小方差。只要使用了一个合理的、正确的替代模型,最大似然法可以推导出一个很好的进化树结果。 由于最大似然法的分析过程需要耗费较多的时间,针对这种情况,发展出了许多优化的可以加快最大似然法寻找最优树的搜索方法,如启发式搜索,分枝交换搜索等。最大似然法具有坚实的统计学理论基础,充分的使用了分析序列中的信息资源,只要采用了合理的替代模型,可以得出很好的进化树分析结果。 构建ML树可以使用PAUP或者PHYLIP来。BioEdit也可以。BioEdit集成了一些PHYLIP的程序,用来构建进化树。

156
可靠性分析:自展法(Bootstrap Method)
构建了进化树之后,需要对其进行评估,即进行可靠性分析:通常采用自展法进行分析 是一种统计学方法,计算的基本方法是:从整个序列的碱基(氨基酸)中随机取出一般,剩下的一半随机补齐,组成一个新的序列。这样,一个序列就可以生成多个序列;一个多序列组就可以变成许多个序列组。根据某种算法,每个多序列组都可以生成一个进化树。将生成的许多个进化树进行比较,按照多数规则(majority-rule),最终将得到一个“逼真”的进化树。 重建一系列与原数据同样大小的取样数据,核酸序列一般会重复500或1000次。 评估进化树的方法。 Bootstrap几乎是一个必须的选项。一般Bootstrap的值>70,则认为构建的进化树较为可靠。如果Bootstrap的值太低,则有可能进化树的拓扑结构有错误,进化树是不可靠的。
中随机取出一般,剩下的一半随机补齐,组成一个新的序列。这样,一个序列就可以生成多个序列;一个多序列组就可以变成许多个序列组。根据某种算法,每个多序列组都可以生成一个进化树。将生成的许多个进化树进行比较,按照多数规则(majority-rule),最终将得到一个 逼真 的进化树。 重建一系列与原数据同样大小的取样数据,核酸序列一般会重复500或1000次。 评估进化树的方法。 Bootstrap几乎是一个必须的选项。一般Bootstrap的值>70,则认为构建的进化树较为可靠。如果Bootstrap的值太低,则有可能进化树的拓扑结构有错误,进化树是不可靠的。")
157
6.4 常用分子进化与系统发育分析的软件 软件名称 网址 说明 PHYLIP (免费的、集成的进化分析工具)
It includes programs to carry out parsimony, distance matrix methods, maximum likelihood, and other methods on a variety of types of data, including DNA and RNA sequences, protein sequences, restriction sites, 0/1 discrete characters data, gene frequencies, continuous characters and distance matrices. (发布最广、用户最多的建树软件) PAUP (商业软件,集成的进化分析工具) It includes parsimony, distance matrix, invariants, and maximum likelihood methods and many indices and statistical tests.(国际上最通用的建树软件之一) Tree of Life (较快的ML建树工具 ) Arizona大学开发的软件 MEGA (图形化、集成的进化分析工具,不包括ML法) 美国宾州州立大学Masatoshi Nei开发(It carries out parsimony, distance matrix and likelihood methods for molecular data.)
 PAUP. (商业软件,集成的进化分析工具) It includes parsimony, distance matrix, invariants, and maximum likelihood methods and many indices and statistical tests.(国际上最通用的建树软件之一) Tree of Life. (较快的ML建树工具 ) Arizona大学开发的软件. MEGA. (图形化、集成的进化分析工具,不包括ML法) 美国宾州州立大学Masatoshi Nei开发(It carries out parsimony, distance matrix and likelihood methods for molecular data.)")
158
软件名称 网址 说明 MOLPHY(ML法建树) 日本国立统计数理研究所开发。(Carrying out maximum likelihood inference of phylogenies for either nucleotide sequences or protein sequences.) PAML (ML建树工具 ) 英国伦敦学院Z. H. YANG开发。(A package of programs for the ML analysis of nucleotide or protein sequences.) PUZZLE ftp://fx.zi.biologie.uni-muenchen.de/pub/puzzle 应用Quarter puzzling方法(一种最大简约法)构建系统发育树 TreeView (进化树显示工具) A program for displaying trees on Apple Macs and Windows PCs. It can draw rooted and unrooted trees, display bootstrap values, and supports the native font and graphics file formats of both Macs and PCs. phylogeny EBI的系统发育树分析软件
 PAML. (ML建树工具 ) 英国伦敦学院Z. H. YANG开发。(A package of programs for the ML analysis of nucleotide or protein sequences.) PUZZLE. ftp://fx.zi.biologie.uni-muenchen.de/pub/puzzle. 应用Quarter puzzling方法(一种最大简约法)构建系统发育树. TreeView. (进化树显示工具) A program for displaying trees on Apple Macs and Windows PCs. It can draw rooted and unrooted trees, display bootstrap values, and supports the native font and graphics file formats of both Macs and PCs. phylogeny. EBI的系统发育树分析软件.")
159
MEGA软件的使用(现在有5.10的版本) www.megasoftware.net/
MEGA软件为初学者的首选。虽然多序列比对工具ClustalW/X自带了一个NJ的建树程序,但是该程序只有p-distance模型,而且构建的树不够准确,一般不用来构建进化树。因此之前的多序列比对步骤,常用Clustal软件进行。

160
例:PKA(蛋白激酶A)家族的系统发育分析
Kinase.coma UniProtb S. Cerevisiae TPK1 KAPA_YEAST TPK2 KAPB_YEAST TPK3 KAPC_YEAST C. elegans kin-1 KAPC_CAEEL D. melanogaster Pka-C1 KAPC_DROME Pka-C2 KDC1_DROME PKA-C3 KDC2_DROME CG12069 Q9VA47_DROME H. sapiens PKACa Q32P54_HUMAN PKACb KAPCB_HUMAN PKACg Q5VZ02_HUMAN PRKX PRKX_HUMAN PRKY PRKY_HUMAN S. Cerevisiae酿酒酵母 C. Elegans秀丽隐杆线虫 D. Melanogaster果蝇

161
所下载的序列都是FASTA格式,并储存于同一个文件中。用Clustal软件进行多序列比对。

162
首先要将多序列比对后的.aln文件转换成MEGA文件格式
MEGA 系列软件用于检验和分析DNA、蛋白质序列的演化。 以下讲的是4.0的版本。5.0版本的mega,大家可以自学。不难。

163
选择序列类型:蛋白质序列

164
选择方法和模型:如NJ法构建进化树

165
概率统计模型:泊松校正模型( Poisson correction)(如果是核酸序列,则选择Kimura 2-parameter)
如果使用MEGA进行分析,选项中有一项是“Gaps/Missing Data”,一般选择“Pairwise Deletion”。其他多数的选项保持缺省的参数。

166
检验:Bootstrap。次数最好大于100。
随机种子可随意设定,不影响结果

167
PKA亚家族的系统发育树 输出的结果中有两棵树,一个是原始树,一个是bootstrap验证过的一致树。
当然还可以有不同的树型、长度、宽度等的选择。 计算基因分化的年代。这个一般需要知道物种的核苷酸替代率。常见物种的核苷酸替代率需要查找相关的文献。 外类群 数字表示bootstrap验证中该树枝可信度的百分比。

168
7 多态性分析

169
用DnaSP软件进行多态性分析 1:根据测序峰图,将双向测序回来的序列删除前后两端不稳定的序列后拼接成一条完整序列。
2:将所有的序列编辑,统一到同一个长度,若是编码蛋白质的基因,则须将序列处理成同一个ORF,然后存储在同一个文本文件中(可将txt的后缀名改成fasta)。 3:用DnaSP软件读取文件(FASTA, MEGA, NBRF/PIR,NEXUS and PHYLIP等格式),出现一个小窗口,描述了所打开序列数据的总的核苷酸数目,序列数目等。将这个窗口关闭。 计算各个群体的多态性(包括单倍体型、变异位点、简约位点等)。从群体的单倍体型的多寡分析遗传多样性。
。 3:用DnaSP软件读取文件(FASTA, MEGA, NBRF/PIR,NEXUS and PHYLIP等格式),出现一个小窗口,描述了所打开序列数据的总的核苷酸数目,序列数目等。将这个窗口关闭。 计算各个群体的多态性(包括单倍体型、变异位点、简约位点等)。从群体的单倍体型的多寡分析遗传多样性。")
170
打开fasta , MEGA, NBRF/PIR,NEXUS and PHYLIP等格式文件
DnaSP软件界面。 出现一个小窗口,描述了所打开序列数据的总的核苷酸数目,序列数目等(默认为常染色体提,二倍体型。如果不是,须在菜单栏“data”选择“format”进行修改)。将这个窗口关闭。
。将这个窗口关闭。")
171
菜单栏的“Display”中选择”data info”可查看序列信息,选择“view data”,可查看比对的序列
菜单栏中“Analysis”选择“DNA polymorphism”可查看比对序列的DNA多态性。其他选择各有用处,如群体之间的DNA多态性等。

172
显示的结果中包含有我们所需的核苷酸多样性Pi,单倍体型多样性Hd

173
summary 如何下载数据库中的核酸序列、蛋白质序列(fasta格式) 如何确定序列的ORF?
如何用Primer Primier设计引物? 测序结果如何判断? 如何将多个序列拼接成一条序列? 如何利用EST数据通过电子克隆的方法获得新基因? 如何将所得序列提交给数据库? 如何用Blast在线程序将序列与数据库中的其他序列进行比对?比对结果如何分析? 如何用ClustalX软件进行多序列比对? 如何用Mega软件建立分子进化树? 如何用DnaSP软件进行群体多样性分析?

Similar presentations



,必须在活细胞内寄生并以复 制方式增殖的非细胞型微生物。 生物病毒微生物 原指一种动物来源的 毒素。 “virus” 一词源于 拉丁文。 病毒能增殖、遗传和演化,因而 具 有生命最基本的特征。>")


















