Download presentation
1
Chapter 3 酶 (Enzyme)
")
2
Section 1 酶 的 基 本 性 质 一、酶是生物催化剂
酶是生物细胞产生的、以蛋白质为主要成分的、能加快化学反应速度、使之迅速达到平衡、但不改变反应的平衡常数的生物催化剂。酶的催化活性受多种因素调节控制。 二、酶能加快细胞内的化学反应速度 按照能量学的观点,一个化学反应并不是从反应物直接向产物生成的方向进行的。反应的发生,即产物的生成取决于过渡态或转换态(transition state)的能量状况。只有当反应物在空间定向上有利于反应以及反应物达到转换态所需的能量(即是否处在化学键的断裂或形成所需的范围内)时,反应才能发生。很显然, 转换态越能有效的达到,反应的速度就越大。如图3—1所示,反应物从基态转变成转换态的能量差值称为活化能(△G‡)。毫无例外, 每一种化学反应都有一个△G‡。
的能量状况。只有当反应物在空间定向上有利于反应以及反应物达到转换态所需的能量(即是否处在化学键的断裂或形成所需的范围内)时,反应才能发生。很显然, 转换态越能有效的达到,反应的速度就越大。如图3—1所示,反应物从基态转变成转换态的能量差值称为活化能(△G‡)。毫无例外, 每一种化学反应都有一个△G‡。")
3
根据转换态的理论,一种催化剂的作用在于以某种途径降低反应的活化能而增高化学反应速度。因此,在有催化剂存在的情况下,转换态能比较有效地达到,从而增高反应的速度。典型的酶促反应速度比无催化剂存在的反应要高出106~1012倍。 三、酶不能改变化学反应的平衡 反应速度和反应平衡之间有着重要的差别。催化剂的功能是增高反应的速度,而不会改变反应的平衡。酶的存在,象其他催化剂一样,只能降低达到转换态所需的活化能而不能改变反应的平衡,它唯一的作用是加速反应物和产物的相互转化。在反应中,酶不会被消耗,平衡点不会改变,但达到平衡的速度则要快得多。反应的平衡与△G0′(产物与反应物所固有的标准自由能之差)有着密切的关系,但反应速度则与△G‡有关。 四、酶的催化反应具有专一性 酶对所催化的底物具有不同程度的选择性。某一种酶往往只能对某一类物质起作用,或者只对某一种物质起催化作用。一般无机催化剂对它们所作用的底物没有这种严格的选择性。
有着密切的关系,但反应速度则与△G‡有关。 四、酶的催化反应具有专一性. 酶对所催化的底物具有不同程度的选择性。某一种酶往往只能对某一类物质起作用,或者只对某一种物质起催化作用。一般无机催化剂对它们所作用的底物没有这种严格的选择性。")
4
五、酶的组成 1.酶的组成 2.酶的活性中心、结合部位和催化部位 ●由单纯蛋白质所构成的酶
●由蛋白质和其他成分构成的酶:除了蛋白质成分(即酶蛋白apoenzyme)外,还含有一些对热稳定的非蛋白质小分子物质(即辅助因子cofactor)。辅助因子分辅酶和辅基(coenzyme & prosthetic group),它们是这类酶的催化活性的必要条件,缺少它们,酶的催化作用就会消失。 2.酶的活性中心、结合部位和催化部位 酶的活性中心通常是指酶分子上直接与底物结合并与催化作用直接有关的部位,是由酶分子上的某些氨基酸残基的侧链基团共同构成。酶的活性中心可分两部分:参与和底物结合的结合部位(binding site) 及直接参与催化反应的催化部位(catalytic site) 。两者对酶活性都是必须的,前者决定酶的专一性,后者决定酶的催化能力。
外,还含有一些对热稳定的非蛋白质小分子物质(即辅助因子cofactor)。辅助因子分辅酶和辅基(coenzyme & prosthetic group),它们是这类酶的催化活性的必要条件,缺少它们,酶的催化作用就会消失。 2.酶的活性中心、结合部位和催化部位. 酶的活性中心通常是指酶分子上直接与底物结合并与催化作用直接有关的部位,是由酶分子上的某些氨基酸残基的侧链基团共同构成。酶的活性中心可分两部分:参与和底物结合的结合部位(binding site) 及直接参与催化反应的催化部位(catalytic site) 。两者对酶活性都是必须的,前者决定酶的专一性,后者决定酶的催化能力。")
5
六、某些RNA具有催化活性 长期以来,人们都认为所有的酶都是蛋白质.但是近20年来,越来越多的例子表明某些RNA分子也是生物催化剂。这些具有催化活性的RNA,叫做Ribozyme,意即酶活性RNA。 例如,RNase P是一种与tRNA前体加工成tRNA反应有关的酶。S.Altman发现,在体外,单独的蛋白质组分不能催化tRNA前体转变成tRNA,而RNA组分在适当的条件下能完成这种转变反应。另外一个例子是T.Cech发现四膜虫26S rRNA前体中的插入顺序的切除与两端片段的正确拼接不需要蛋白质的催化,是一种自我剪接的过程。该过程需要鸟苷或者鸟苷酸的存在.在体内,这种剪接方式可能只进行一次,但在体外,他象一种真正的酶一样能作用多次。 H.F.Noller及其同事发现蛋白质合成时,肽键的形成很可能是50S核糖体亚基催化的。催化这一反应的酶叫做肽基转移酶。当把所有的蛋白质组分从50S核糖体亚基中去除后,23S rRNA能催化这一肽键的形成反应(图3—2).
.")
6
七、催化抗体---抗体酶 抗体是免疫球蛋白。催化抗体(catalytic antibodies)又叫做抗体酶(abzymes)。象其它抗体一样, 抗体酶也是一种生物对叫做抗原(antigen)的某种外源分子作出应答的产物, 只是这种抗原分子被有目的地改造成某种反应的转换态中间物。推理是:一种能专一同某反应的转换态中间物结合的蛋白质,必定能启动正常反应物进入到活泼的转换态构象。因此, 一种催化抗体便能促使它的底物形成转换态构象,从而加速这一反应。常规酶最显著的催化效力是因为它们对所催化的反应转换态中间物有很高的亲和力,因而抗体酶同它的专一性底物(抗原)必定有很高亲和力。
又叫做抗体酶(abzymes)。象其它抗体一样, 抗体酶也是一种生物对叫做抗原(antigen)的某种外源分子作出应答的产物, 只是这种抗原分子被有目的地改造成某种反应的转换态中间物。推理是:一种能专一同某反应的转换态中间物结合的蛋白质,必定能启动正常反应物进入到活泼的转换态构象。因此, 一种催化抗体便能促使它的底物形成转换态构象,从而加速这一反应。常规酶最显著的催化效力是因为它们对所催化的反应转换态中间物有很高的亲和力,因而抗体酶同它的专一性底物(抗原)必定有很高亲和力。")
7
Section 2. 酶的命名与分类 一. 命名原则 1.习惯命名 例如:乙醇脱氢酶 2.系统命名
1.习惯命名 例如:乙醇脱氢酶 2.系统命名 1).应标明酶的所有底物及催化的性质,并用: 符号把底物分开。例如:乙醇:NAD+脱氢酶. 2).不管催化正反应还是逆反应,一般都用同一名称。 3).如果所催化的反应包含二种变化,则尽可能用同一种变化 来表示,另一个功能附后。 例如:L-谷氨脱氢酶催化的反应是:(图3-3) 谷氨酸:NAD+脱氢酶(脱氨)。
.应标明酶的所有底物及催化的性质,并用: 符号把底物分开。例如:乙醇:NAD+脱氢酶. 2).不管催化正反应还是逆反应,一般都用同一名称。 3).如果所催化的反应包含二种变化,则尽可能用同一种变化. 来表示,另一个功能附后。 例如:L-谷氨脱氢酶催化的反应是:(图3-3) 谷氨酸:NAD+脱氢酶(脱氨)。")
8
二.酶的分类(图3-3) 1.氧化还原酶类: 催化氧化还原反应:A·2H + B → A + B· 可分为 需氧脱氢酶、氧化酶和不需氧脱氢酶。 2.转移酶类(transferanses): 催化功能基团的转移反应:AB + C ←→A + BC 3.水解酶类(hydrolases): 催化水解反应:AB + HOH →AOH + BH 4.裂合酶类(lyases):催化底物裂解反应,并产生双键,或其 逆反应,将一个基团加到双键上:A·B ←→ A + B 例如: (图3-4) 5.异构酶类(isomerases):催化各种同分异构体的相互转变: A ←→ B 如异构酶和消旋酶催化的反应。 6.合成酶(连接酶)类(ligases):这类酶催化一切必须与ATP分解相联,并由二种物质合成一种物质的反应: A + B + ATP → A·B + ADP + Pi(或 AMP + PPi) 例如谷氨酰胺合成酶催化的反应:(图3-3)
: 催化水解反应:AB + HOH →AOH + BH. 4.裂合酶类(lyases):催化底物裂解反应,并产生双键,或其. 逆反应,将一个基团加到双键上:A·B ←→ A + B 例如: (图3-4) 5.异构酶类(isomerases):催化各种同分异构体的相互转变: A ←→ B 如异构酶和消旋酶催化的反应。 6.合成酶(连接酶)类(ligases):这类酶催化一切必须与ATP分解相联,并由二种物质合成一种物质的反应: A + B + ATP → A·B + ADP + Pi(或 AMP + PPi) 例如谷氨酰胺合成酶催化的反应:(图3-3)")
9
Section 3 酶反应动力学 一、酶活力与酶的反应速度 要检查酶的存在及含量, 不能直接用重量或体积来表示, 要根据酶引起的某一特定的化学反应的能力来表示, 即用酶的活力来表示. 酶活力的高低是研究酶的特性、进行酶制剂的生产及应用时的一项不可缺少的指标.酶活力又叫酶活性(activity)是指酶催化一定化学反应的能力. 1.酶的活力测定 酶反应速度可以用单位时间内、单位体积中底物的减少量或产物的生成量表示, 即用: -d[s]/d[t] 或 d[p]/d[t] 表示. 研究酶反应速度或测定酶的活性应以酶促反应的初速度(initial rate)为准.初速度是指在酶浓度底物浓度一定的条件下, 在最初一段时间内, 产物的浓度随时间的增加而成比例的增加时的速度(图3-6)。
是指酶催化一定化学反应的能力. 1.酶的活力测定. 酶反应速度可以用单位时间内、单位体积中底物的减少量或产物的生成量表示, 即用: -d[s]/d[t] 或 d[p]/d[t] 表示. 研究酶反应速度或测定酶的活性应以酶促反应的初速度(initial rate)为准.初速度是指在酶浓度底物浓度一定的条件下, 在最初一段时间内, 产物的浓度随时间的增加而成比例的增加时的速度(图3-6)。")
10
2. 酶的活力单位. 国际酶学委员会规定, 一个酶活力单位(unit,u) 是指在25℃下, 其它条件如pH及底物浓度均采用最适条件, 在1分钟内转化1μmol底物的酶量(或转化1μmol底物的有关基团的酶量).但是,酶的活力单位常根据实际需要来规定。 3、酶的比活力(specific activity) 比活力是指每mg蛋白质所具有的活力单位数, 即活力单位数/mg蛋白质. 在酶的纯化过程中, 每步纯化后都要测定酶的比活力, 比活力高, 表明酶的纯度高。 酶的纯度是用比活力表示的: 比活力 = 活力单位数/mg蛋白 = 总活力单位数/总mg蛋白.
 比活力是指每mg蛋白质所具有的活力单位数, 即活力单位数/mg蛋白质. 在酶的纯化过程中, 每步纯化后都要测定酶的比活力, 比活力高, 表明酶的纯度高。 酶的纯度是用比活力表示的: 比活力 = 活力单位数/mg蛋白 = 总活力单位数/总mg蛋白.")
11
例如, 某酶经过四个纯化步骤后, 获得如下的数据:
纯化步骤: Ⅰ Ⅱ Ⅲ Ⅳ 总活力: 总蛋白: 比活力: / /10 3/5 2/2 在提纯中,总活性在减少,总蛋白也在减少,但比活性在提高。 此外, 在酶的纯化工作中还要计算两个具有实际意义的项目, 即纯化倍数和回收率(产量%): 纯化倍数 = 每次比活力/第一次比活力 回收率(产量%)=(每次的总活力/第一次总活力)×100 作业:根据所学知识设计撰写某种酶分离纯化的技术报告。
: 纯化倍数 = 每次比活力/第一次比活力. 回收率(产量%)=(每次的总活力/第一次总活力)×100. 作业:根据所学知识设计撰写某种酶分离纯化的技术报告。")
12
二、酶反应的基本动力学 酶动力学研究的是酶催化反应的速度,描述酶作为催化剂的生物学作用, 描述酶是怎样完成它们所催化的反应。由于酶的最基本特性就是加快生物化学反应速度,因此,所有酶的催化反应速度以及它们的催化效率都是可以定量的。这就是酶促反应动力学研究的基本内容。 1. 底物浓度对酶促反应速度的影响. 底物浓度对酶促反应速度的影响如图3-7所示。

13
1) 中间产物学说 酶动力学研究始于1902年,当时,A.Brown用酵母β-呋喃果糖糖苷酶对蔗糖进行水解。他发现当蔗糖的浓度比酶的浓度高许多时,反应速度不再取决于蔗糖的浓度,即是说,反应速度相对于蔗糖来说是零级反应。因此,他提出,蔗糖水解的总反应是由两个基本反应构成,第一步是底物与酶形成一种复合物,第二步是这种中间物生成产物并释放出酶: k k2 E + S ←→ ES → P + E k-1 这里,E、S、ES和P分别代表酶、底、酶-底物复合物和产物。 中间产物学说认为, 当酶催化某个反应时, 酶和底物先结合形成一个中间复合物, 然后中间复合物再分解, 生成产物并释放出酶。一般以产物的生成速度代表整个酶催化反应速度,而产物的生成取决于中间物的浓度. 因此, 整个酶促反应的速度取决于中间物的浓度.

14
2) 酶促反应的基本方程----米氏方程 根据中间产物学说的原理、并假定第二步反应是不可逆的,于是推导出了米氏方程: ν=Vmax ·[S]/(Km +[S]) v代表底物浓度为[S]时的反应速度,Vmax代表最大反应速度, Km为米氏常数,等于(k-1+k2)/k1 . 米氏方程表明了当Km和Vmax都已知时,酶反应速度与底物浓度间的关系。
![2) 酶促反应的基本方程----米氏方程 根据中间产物学说的原理、并假定第二步反应是不可逆的,于是推导出了米氏方程: ν=Vmax ·[S]/(Km +[S]) v代表底物浓度为[S]时的反应速度,Vmax代表最大反应速度,](http://slidesplayer.com/slide/11480556/62/images/14/2%29+%E9%85%B6%E4%BF%83%E5%8F%8D%E5%BA%94%E7%9A%84%E5%9F%BA%E6%9C%AC%E6%96%B9%E7%A8%8B----%E7%B1%B3%E6%B0%8F%E6%96%B9%E7%A8%8B+%E6%A0%B9%E6%8D%AE%E4%B8%AD%E9%97%B4%E4%BA%A7%E7%89%A9%E5%AD%A6%E8%AF%B4%E7%9A%84%E5%8E%9F%E7%90%86%E3%80%81%E5%B9%B6%E5%81%87%E5%AE%9A%E7%AC%AC%E4%BA%8C%E6%AD%A5%E5%8F%8D%E5%BA%94%E6%98%AF%E4%B8%8D%E5%8F%AF%E9%80%86%E7%9A%84%EF%BC%8C%E4%BA%8E%E6%98%AF%E6%8E%A8%E5%AF%BC%E5%87%BA%E4%BA%86%E7%B1%B3%E6%B0%8F%E6%96%B9%E7%A8%8B%EF%BC%9A+%CE%BD%EF%BC%9DVmax+%C2%B7%EF%BC%BBS%EF%BC%BD%EF%BC%8F%EF%BC%88Km+%2B%EF%BC%BBS%EF%BC%BD%EF%BC%89+v%E4%BB%A3%E8%A1%A8%E5%BA%95%E7%89%A9%E6%B5%93%E5%BA%A6%E4%B8%BA%EF%BC%BBS%EF%BC%BD%E6%97%B6%E7%9A%84%E5%8F%8D%E5%BA%94%E9%80%9F%E5%BA%A6%EF%BC%8CVmax%E4%BB%A3%E8%A1%A8%E6%9C%80%E5%A4%A7%E5%8F%8D%E5%BA%94%E9%80%9F%E5%BA%A6%EF%BC%8C.jpg "Km为米氏常数,等于(k-1+k2)/k1 . 米氏方程表明了当Km和Vmax都已知时,酶反应速度与底物浓度间的关系。")
15
(-1+ cat )[ES]= 1([E]total-[ES])[S] (4.4)
4.6.1米氏方程的推导 假定存在一个时间周期,在此期间 S 结合 E的速度与 ES 解离的速度相同。此时[ES]是个恒定值,因为 ES 解离为 E+S 或 E+P 的速度等于由 E+S 形成 ES 的速度。由E+S 形成 ES的速度取决于游离酶的浓度,即等于[E]total-[ES]。可写成: (-1+ cat )[ES]= 1([E]total-[ES])[S] (4.4) 整理方程,将速度常数集中在一边,并定义为米氏常数Km。 (-1+ cat ) ([E]total-[ES])[S] —————— = Km = ———————— (4.5) [ES] 从方程(4.5):Km[ES]=([E]total-[ES])[S] [ES](Km+[S])=[E]total[S]
![(-1+ cat )[ES]= 1([E]total-[ES])[S] (4.4)](http://slidesplayer.com/slide/11480556/62/images/15/%EF%BC%88%EF%81%AB%EF%BC%8D1%EF%BC%8B%EF%81%AB+cat+%EF%BC%89%5BES%5D%EF%BC%9D%EF%81%AB+1%EF%BC%88%5BE%5Dtotal%EF%BC%8D%5BES%5D%EF%BC%89%5BS%5D+%EF%BC%884.4%EF%BC%89.jpg "4.6.1米氏方程的推导. 假定存在一个时间周期,在此期间 S 结合 E的速度与 ES 解离的速度相同。此时[ES]是个恒定值,因为 ES 解离为 E+S 或 E+P 的速度等于由 E+S 形成 ES 的速度。由E+S 形成 ES的速度取决于游离酶的浓度,即等于[E]total-[ES]。可写成: (-1+ cat )[ES]= 1([E]total-[ES])[S] (4.4) 整理方程,将速度常数集中在一边,并定义为米氏常数Km。 (-1+ cat ) ([E]total-[ES])[S] —————— = Km = ———————— (4.5) 1 [ES] 从方程(4.5):Km[ES]=([E]total-[ES])[S] [ES](Km+[S])=[E]total[S]")
16
由于酶促反应速度取决于ES转换为E+P的速度。
[E]total[S] [ES]= ———— (4.6) Km+[S] 由于酶促反应速度取决于ES转换为E+P的速度。 = cat[ES] (4.7) 将方程(4.6)代入(4.7), cat [E]total[S] = —————— (4.8) 因为 cat [E]total=Vmax,代入方程(4.8),就得到了米氏方程。 Vmax[S] =—————— (4.9)
 Km+[S] 由于酶促反应速度取决于ES转换为E+P的速度。 = cat[ES] (4.7) 将方程(4.6)代入(4.7), cat [E]total[S] = —————— (4.8) 因为 cat [E]total=Vmax,代入方程(4.8),就得到了米氏方程。 Vmax[S] =—————— (4.9)")
17
-1 / 1 ,这是 ES 解离为E+S的平衡常数。
(-1+ cat ) —————— = Km 1 如果 cat比 1或-1小得多, cat可以忽略,Km就变成了 -1 / 1 ,这是 ES 解离为E+S的平衡常数。 同样Michaelis和Menten假定ES与E+S处于快速的平衡中, Km就是 ES 的解离常数。因此 Km是 E 对 S 的亲和力的量度。如果 Km的值越小,表明 E 与 S 结合的越紧密。 如果一个酶可作用几个底物,例如己糖激酶对葡萄糖的Km值为1.5×10-4;对果糖的Km值为1.5×10-3 ,显然酶对葡萄糖的亲和性更高,葡萄糖就称之为己糖激酶的天然底物。
 —————— = Km. 1. 如果 cat比 1或-1小得多, cat可以忽略,Km就变成了. -1 / 1 ,这是 ES 解离为E+S的平衡常数。 同样Michaelis和Menten假定ES与E+S处于快速的平衡中, Km就是 ES 的解离常数。因此 Km是 E 对 S 的亲和力的量度。如果 Km的值越小,表明 E 与 S 结合的越紧密。 如果一个酶可作用几个底物,例如己糖激酶对葡萄糖的Km值为1.5×10-4;对果糖的Km值为1.5×10-3 ,显然酶对葡萄糖的亲和性更高,葡萄糖就称之为己糖激酶的天然底物。")
18
3) Km和Vmax的求解: 当反应速度达到最大反应速度一半时,即V = Vmax/2 时,得到: Vmax/2 = Vmax[S]/(Km +[S]), 即 Km=[S] 可见,Km 值本身的含义是:酶反应速度达到最大反应速度一半时的底物浓度。∴Km 的单位与底物浓度的单位一致。 为了准确地求出Km ,可把米氏方程改成直线方程,用图解法求出Km,常用双倒数作图法: 1/v = (Km +[S]) /Vmax[S] 即 1/v = (Km/Vmax)·(1/[S] )+ 1/ Vmax 以1/v对1/[S]作图即可得到如图3-8a所示的结果。
![3) Km和Vmax的求解: 当反应速度达到最大反应速度一半时,即V = Vmax/2 时,得到: Vmax/2 = Vmax[S]/(Km +[S]), 即 Km=[S] 可见,Km 值本身的含义是:酶反应速度达到最大反应速度一半时的底物浓度。∴Km 的单位与底物浓度的单位一致。](http://slidesplayer.com/slide/11480556/62/images/18/3%29+Km%E5%92%8CVmax%E7%9A%84%E6%B1%82%E8%A7%A3%EF%BC%9A+%E5%BD%93%E5%8F%8D%E5%BA%94%E9%80%9F%E5%BA%A6%E8%BE%BE%E5%88%B0%E6%9C%80%E5%A4%A7%E5%8F%8D%E5%BA%94%E9%80%9F%E5%BA%A6%E4%B8%80%E5%8D%8A%E6%97%B6%EF%BC%8C%E5%8D%B3V+%3D+Vmax%EF%BC%8F2+%E6%97%B6%EF%BC%8C%E5%BE%97%E5%88%B0%EF%BC%9A+Vmax%EF%BC%8F2+%3D+Vmax%EF%BC%BBS%EF%BC%BD%EF%BC%8F%28Km+%EF%BC%8B%EF%BC%BBS%EF%BC%BD%29%2C+%E5%8D%B3+Km%EF%BC%9D%EF%BC%BBS%EF%BC%BD+%E5%8F%AF%E8%A7%81%EF%BC%8CKm+%E5%80%BC%E6%9C%AC%E8%BA%AB%E7%9A%84%E5%90%AB%E4%B9%89%E6%98%AF%EF%BC%9A%E9%85%B6%E5%8F%8D%E5%BA%94%E9%80%9F%E5%BA%A6%E8%BE%BE%E5%88%B0%E6%9C%80%E5%A4%A7%E5%8F%8D%E5%BA%94%E9%80%9F%E5%BA%A6%E4%B8%80%E5%8D%8A%E6%97%B6%E7%9A%84%E5%BA%95%E7%89%A9%E6%B5%93%E5%BA%A6%E3%80%82%E2%88%B4Km+%E7%9A%84%E5%8D%95%E4%BD%8D%E4%B8%8E%E5%BA%95%E7%89%A9%E6%B5%93%E5%BA%A6%E7%9A%84%E5%8D%95%E4%BD%8D%E4%B8%80%E8%87%B4%E3%80%82.jpg "为了准确地求出Km ,可把米氏方程改成直线方程,用图解法求出Km,常用双倒数作图法: 1/v = (Km +[S]) /Vmax[S] 即 1/v = (Km/Vmax)·(1/[S] )+ 1/ Vmax. 以1/v对1/[S]作图即可得到如图3-8a所示的结果。")
19
4)Km 的意义 ▼Km是酶的一个基本的特征性常数,只与酶的性质有关,与酶的浓度无关,但与pH、温度等因素有关;不同酶有不同的Km。 ▼如果一种酶可作用于几种底物,就有几个不同的Km,Km值小的底物一般作为该酶的最适底物或天然底物。 ▼如果k2<<k-1时,k2可忽略不计,那么, Km=k-1/k1=1/k亲和力=ks (1) 这里,ks代表ES解离的平衡常数。这个假定的提出,仅仅针对 E+S=ES,此时, Km 的大小(或 1/Km的大小),可近似地说明酶与底物亲和力的大小。 ▼从Km 的大小可知正确测定酶活力时所需的底物浓度。如果要求反应速度达到Vmax的90%,底物浓度应为: 90%=100%[S]/(Km +[S]) , [S]= 9 Km ; 而要达到最大反应速度的99%,底物浓度应为: 99% = 100%[S]/(Km +[S]) , [S]= 99Km
 这里,ks代表ES解离的平衡常数。这个假定的提出,仅仅针对 E+S=ES,此时, Km 的大小(或 1/Km的大小),可近似地说明酶与底物亲和力的大小。 ▼从Km 的大小可知正确测定酶活力时所需的底物浓度。如果要求反应速度达到Vmax的90%,底物浓度应为: 90%=100%[S]/(Km +[S]) , [S]= 9 Km ; 而要达到最大反应速度的99%,底物浓度应为: 99% = 100%[S]/(Km +[S]) , [S]= 99Km.")
20
5)酶的转换数 酶的转换数(turnover number,即kcat)又称为酶的摩尔活性或者催化中心活性, 用来表示酶的催化效力(catalytic power), 其定义为: 每摩尔的酶(单体酶)或每摩尔的酶活性中心(含有多个活性中心的酶)在单位时间内转化底物的摩尔数. 假定反应混合物中的酶浓度([ET])是已知道, 在饱和的[S]下, v = Vmax = k2[ET],Vmax揭示了转换数。 因此, k2 = Vmax/[ET] = kcat 6)kcat/Km. 动力学参数kcat和Km对于不同酶的研究和比较具有普遍性用途。每一种酶都有最适合的kcat和Km值, 反映出细胞环境、体内被酶正常遭遇到的底物浓度以及被催化的化学反应等。 不同酶的催化效率的比较需要选择适当的参数。常数kcat并不令人满意。两种催化不同反应的酶可能有相同的kcat,然而它们非催化的速度也许是不同的。
是已知道, 在饱和的[S]下, v = Vmax = k2[ET],Vmax揭示了转换数。 因此, k2 = Vmax/[ET] = kcat. 6)kcat/Km. 动力学参数kcat和Km对于不同酶的研究和比较具有普遍性用途。每一种酶都有最适合的kcat和Km值, 反映出细胞环境、体内被酶正常遭遇到的底物浓度以及被催化的化学反应等。 不同酶的催化效率的比较需要选择适当的参数。常数kcat并不令人满意。两种催化不同反应的酶可能有相同的kcat,然而它们非催化的速度也许是不同的。")
21
因此, 由酶所引起的速度提高可能有很大的差别, 而且kcat反映的是一种酶当它被饱和时的性质。在低[S]下,它则失去意义。常数Km由于它自身的原因也不很令人满意。由于当v = Vmax/2时, Km =[S], Km必须对细胞内的正常[S]有某种关系。一种能与以很低的浓度存在于细胞中的底物作用的酶,倾向于有较低的Km(与该酶处在正常丰度的底物下相比)。对于讨论催化效率来说, 最有用的参数应该包含kcat和Km两者。根据米氏方程的推导, Vmax = kcat[ET],代入米-曼氏方程得到 v = kcat[ET][S]/(Km +[S]) 当[S]<< Km时, 游离酶的浓度[E]大体上接近[ET], 于是就得到 v = kcat[ET][S]/Km = (kcat/Km)·[E][S] 在这种情况下, v取决于两种反应物E和S的浓度, 因而这是一种二级反应, 常数kcat/Km是二级速度常数。kcat/Km一般认为是用来比较催化效率的最好的动力学参数。对于kcat/Km来说,它有一个上限, 由E和S在水溶液中扩散相遇所限制. 因为一种酶的催化效率不可能超过E和S结合形成ES的扩散控制限制.这种扩散控制限制(diffusion-controlled limit)大约是108―109 mol·L-1·S-1。许多酶的kcat/Km值接近这一范围.
![因此, 由酶所引起的速度提高可能有很大的差别, 而且kcat反映的是一种酶当它被饱和时的性质。在低[S]下,它则失去意义。常数Km由于它自身的原因也不很令人满意。由于当v = Vmax/2时, Km =[S], Km必须对细胞内的正常[S]有某种关系。一种能与以很低的浓度存在于细胞中的底物作用的酶,倾向于有较低的Km(与该酶处在正常丰度的底物下相比)。对于讨论催化效率来说, 最有用的参数应该包含kcat和Km两者。根据米氏方程的推导, Vmax = kcat[ET],代入米-曼氏方程得到](http://slidesplayer.com/slide/11480556/62/images/21/%E5%9B%A0%E6%AD%A4%2C+%E7%94%B1%E9%85%B6%E6%89%80%E5%BC%95%E8%B5%B7%E7%9A%84%E9%80%9F%E5%BA%A6%E6%8F%90%E9%AB%98%E5%8F%AF%E8%83%BD%E6%9C%89%E5%BE%88%E5%A4%A7%E7%9A%84%E5%B7%AE%E5%88%AB%2C+%E8%80%8C%E4%B8%94kcat%E5%8F%8D%E6%98%A0%E7%9A%84%E6%98%AF%E4%B8%80%E7%A7%8D%E9%85%B6%E5%BD%93%E5%AE%83%E8%A2%AB%E9%A5%B1%E5%92%8C%E6%97%B6%E7%9A%84%E6%80%A7%E8%B4%A8%E3%80%82%E5%9C%A8%E4%BD%8E%5BS%5D%E4%B8%8B%EF%BC%8C%E5%AE%83%E5%88%99%E5%A4%B1%E5%8E%BB%E6%84%8F%E4%B9%89%E3%80%82%E5%B8%B8%E6%95%B0Km%E7%94%B1%E4%BA%8E%E5%AE%83%E8%87%AA%E8%BA%AB%E7%9A%84%E5%8E%9F%E5%9B%A0%E4%B9%9F%E4%B8%8D%E5%BE%88%E4%BB%A4%E4%BA%BA%E6%BB%A1%E6%84%8F%E3%80%82%E7%94%B1%E4%BA%8E%E5%BD%93v+%EF%BC%9D+Vmax%2F2%E6%97%B6%2C+Km+%EF%BC%9D%5BS%5D%2C+Km%E5%BF%85%E9%A1%BB%E5%AF%B9%E7%BB%86%E8%83%9E%E5%86%85%E7%9A%84%E6%AD%A3%E5%B8%B8%5BS%5D%E6%9C%89%E6%9F%90%E7%A7%8D%E5%85%B3%E7%B3%BB%E3%80%82%E4%B8%80%E7%A7%8D%E8%83%BD%E4%B8%8E%E4%BB%A5%E5%BE%88%E4%BD%8E%E7%9A%84%E6%B5%93%E5%BA%A6%E5%AD%98%E5%9C%A8%E4%BA%8E%E7%BB%86%E8%83%9E%E4%B8%AD%E7%9A%84%E5%BA%95%E7%89%A9%E4%BD%9C%E7%94%A8%E7%9A%84%E9%85%B6%2C%E5%80%BE%E5%90%91%E4%BA%8E%E6%9C%89%E8%BE%83%E4%BD%8E%E7%9A%84Km%28%E4%B8%8E%E8%AF%A5%E9%85%B6%E5%A4%84%E5%9C%A8%E6%AD%A3%E5%B8%B8%E4%B8%B0%E5%BA%A6%E7%9A%84%E5%BA%95%E7%89%A9%E4%B8%8B%E7%9B%B8%E6%AF%94%29%E3%80%82%E5%AF%B9%E4%BA%8E%E8%AE%A8%E8%AE%BA%E5%82%AC%E5%8C%96%E6%95%88%E7%8E%87%E6%9D%A5%E8%AF%B4%2C+%E6%9C%80%E6%9C%89%E7%94%A8%E7%9A%84%E5%8F%82%E6%95%B0%E5%BA%94%E8%AF%A5%E5%8C%85%E5%90%ABkcat%E5%92%8CKm%E4%B8%A4%E8%80%85%E3%80%82%E6%A0%B9%E6%8D%AE%E7%B1%B3%E6%B0%8F%E6%96%B9%E7%A8%8B%E7%9A%84%E6%8E%A8%E5%AF%BC%2C+Vmax+%EF%BC%9D+kcat%5BET%5D%2C%E4%BB%A3%E5%85%A5.jpg "v = kcat[ET][S]/(Km +[S]) 当[S]<< Km时, 游离酶的浓度[E]大体上接近[ET], 于是就得到. v = kcat[ET][S]/Km = (kcat/Km)·[E][S] 在这种情况下, v取决于两种反应物E和S的浓度, 因而这是一种二级反应, 常数kcat/Km是二级速度常数。kcat/Km一般认为是用来比较催化效率的最好的动力学参数。对于kcat/Km来说,它有一个上限, 由E和S在水溶液中扩散相遇所限制. 因为一种酶的催化效率不可能超过E和S结合形成ES的扩散控制限制.这种扩散控制限制(diffusion-controlled limit)大约是108―109 mol·L-1·S-1。许多酶的kcat/Km值接近这一范围.")
22
2.pH对酶促反应的影响 酶只能在一定的pH下才能很好的工作。能表现出最大催化活性的pH, 称作该酶的最适pH(optimum pH).pH对酶反应速度的影响如图3-9所示。 pH影响酶活性的原因: 在酶促反应中, 游离酶、底物及酶-底物复合物都可能受环境pH的改变而影响其解离状态。在最适pH范围内, 酶活性中心基团的解离与底物的解离可能处于最佳结合状态;酶活性中心基团的解离可能最有效地发挥酸-碱催化的效率或它们的亲核性或亲电性.

23
3. 温度对酶促反应的影响 每一种酶在一定条件下, 只有在某一温度下才表现最大活力,这个温度称作该酶的最适温度(optimum temperature).温度影响酶的稳定性,酶只有在一定的温度下才是稳当的。温度对酶反应速度的影响如图3-10所示。 4.酶浓度对酶促反应速度的影响 v = k2 [ET] [S]/(km+[S])
")
24
三.多底物酶促动力学 通常,酶催化反应涉及两个(少数情况下三个)底物。现在我们考虑一个涉及两种底物和两种产物的酶促反应. A + B —→ P + Q 这样的反应叫双底物反应。约60%的已知生物化学反应属于双底物酶促反应。一般来说, 双底物反应以下面两种可能路线进行: 1.顺次反应: 所有底物都必须在反应发生前以及产物释放之前与酶结合的反应称为顺次反应。在反应中,被转移的基团直接从底物A进入到底物B, 生成产物P和Q。这样的反应又叫单置换(取代)反应。顺次反应(sequential reaction)又可分为有序顺次反应(ordered reaction)和随机顺次反应(random reaction)两种机制. ◆有序顺次反应(如图3-11a所示) 这里A和B分别可被说成是先导底物和后随底物,Q是A的产物,最后被释放。A和Q竞争同游离E结合,但A和B则不会(或者Q和B也不会)。依赖烟酰胺腺嘌呤二核苷酸(NAD+和NADP+)的脱氢酶服从有序顺次的双底物反应机制。
反应。顺次反应(sequential reaction)又可分为有序顺次反应(ordered reaction)和随机顺次反应(random reaction)两种机制. ◆有序顺次反应(如图3-11a所示) 这里A和B分别可被说成是先导底物和后随底物,Q是A的产物,最后被释放。A和Q竞争同游离E结合,但A和B则不会(或者Q和B也不会)。依赖烟酰胺腺嘌呤二核苷酸(NAD+和NADP+)的脱氢酶服从有序顺次的双底物反应机制。")
25
◆随机顺次反应(如图3-11b所示) A和B两个底物无论哪个先同酶结合都没关系;同样地,产物P和Q谁先释放也不重要。某些激酶例如肌酸激酶服从随机顺次反应机制,也(可能)有某些脱氢酶反应亦属于此类。 2.乒乓反应 在这种反应中,在所有底物加入之前,一种产物被释放的基团转移反应叫做乒乓反应,如图3-11c所示. 这里, 第一个底物A(相当于P―X)的功能基团(X)被酶从底物上置换,产生第一个产物P和一个稳定的酶形式F(相当于E―X). 在F中, X是紧密地(往往共价地)同酶结合(乒).在反应的第二阶段, X被第二种底物B从酶分子上取代, 生成第二个产物Q(B―X),酶也恢复到最初的形式(乓). 这样的反应因此而叫做双置换(取代)反应. 注意, 在乒乓反应中, 底物A和B并不彼此在酶分子上相遇. 许多酶,包括转氨酶、某些黄素酶都具乒乓反应机制.
的功能基团(X)被酶从底物上置换,产生第一个产物P和一个稳定的酶形式F(相当于E―X). 在F中, X是紧密地(往往共价地)同酶结合(乒).在反应的第二阶段, X被第二种底物B从酶分子上取代, 生成第二个产物Q(B―X),酶也恢复到最初的形式(乓). 这样的反应因此而叫做双置换(取代)反应. 注意, 在乒乓反应中, 底物A和B并不彼此在酶分子上相遇. 许多酶,包括转氨酶、某些黄素酶都具乒乓反应机制.")
26
Section 4 酶的抑制作用 一.酶抑制作用及其类型
一.酶抑制作用及其类型 酶的催化反应速度因某种物质同酶结合后而降低的现象称之为抑制作用(inhibition)。造成抑制作用的物质称为抑制剂(inhibitor). 酶的抑制作用和酶的变性作用是不相同的. 酶抑制作用的类型:根据抑制剂与酶作用的特点, 可将抑制作用分为两大类: 不可逆抑制作用和可逆抑制作用。 二.不可逆抑制作用 这类抑制剂与酶的某些基团以共价键的方式结合。必须通过其他的化学反应才能把抑制剂从酶分子上除去。该抑制作用是因为降低了酶的Vmax,故有时也称作非竞争性抑制作用。不可逆抑制剂有:有机磷、有机汞、有机砷、氰化物、烷化剂等化合物。有机磷化合物能同酶蛋白分子中的或酶活性中心上的ser残基的侧链-OH结合,从而抑制酶活性。例如能抑制酯酶或某些蛋白酶的活性。
。造成抑制作用的物质称为抑制剂(inhibitor). 酶的抑制作用和酶的变性作用是不相同的. 酶抑制作用的类型:根据抑制剂与酶作用的特点, 可将抑制作用分为两大类: 不可逆抑制作用和可逆抑制作用。 二.不可逆抑制作用. 这类抑制剂与酶的某些基团以共价键的方式结合。必须通过其他的化学反应才能把抑制剂从酶分子上除去。该抑制作用是因为降低了酶的Vmax,故有时也称作非竞争性抑制作用。不可逆抑制剂有:有机磷、有机汞、有机砷、氰化物、烷化剂等化合物。有机磷化合物能同酶蛋白分子中的或酶活性中心上的ser残基的侧链-OH结合,从而抑制酶活性。例如能抑制酯酶或某些蛋白酶的活性。")
27
乙酰胆碱酯酶能催化乙酰胆碱水解成胆碱和乙酸:
CH3-C-O-CH2CH2N(CH3)3 —→CH3COOH + HO-CH2CH2N(CH3)3 在二异丙基氟磷酸(DIFP)存在下,抑制该酶的活性(图3-12) 有机汞、有机砷化合物能抑制含巯基的酶的活性。主要是能与-SH结合,使酶失活。例如对氯汞苯甲酸: 酶-SH + Cl-Hg-苯环-COOH → 酶-S-Hg-苯环-COOH + HCl 烷化剂如碘乙酸(ICH2-COOH)、碘乙酰胺(ICH2-CO-NH2)也能抑制含-SH的酶的活性,也能与酶活性中心的His的咪唑基共价结合而使酶失活,氰化物能同含铁卟啉的酶的Fe2+结合,使酶失活。例如细胞色素氧化酶等。 三、可逆抑制作用 这类抑制剂与酶的结合是可逆的,抑制剂结合后可用透析等方法除去,使酶的活性恢复。这类结合往往是非共价的。根据抑制剂及底物与酶的关系,分为:竞争性抑制、反竞争性抑制和非竞争性抑制。
3 —→CH3COOH + HO-CH2CH2N(CH3)3. 在二异丙基氟磷酸(DIFP)存在下,抑制该酶的活性(图3-12) 有机汞、有机砷化合物能抑制含巯基的酶的活性。主要是能与-SH结合,使酶失活。例如对氯汞苯甲酸: 酶-SH + Cl-Hg-苯环-COOH → 酶-S-Hg-苯环-COOH + HCl. 烷化剂如碘乙酸(ICH2-COOH)、碘乙酰胺(ICH2-CO-NH2)也能抑制含-SH的酶的活性,也能与酶活性中心的His的咪唑基共价结合而使酶失活,氰化物能同含铁卟啉的酶的Fe2+结合,使酶失活。例如细胞色素氧化酶等。 三、可逆抑制作用. 这类抑制剂与酶的结合是可逆的,抑制剂结合后可用透析等方法除去,使酶的活性恢复。这类结合往往是非共价的。根据抑制剂及底物与酶的关系,分为:竞争性抑制、反竞争性抑制和非竞争性抑制。")
28
1.竞争性抑制剂只同自由酶结合 竞争性抑制剂(competitive inhibition )与底物竞争酶的活性中心,阻止底物与酶的结合,这类抑制作用称为竞争性抑制. 在正常情况下: E + S →ES → P + E 当有抑制剂存在时:E + I → EI ; EI不能转变为产物,实际上降低了酶的浓度,因而使反应速度下降。 这类抑制剂的结构往往与酶所作用的底物的结构相似,其抑制程度主要取决于抑制剂和底物各自的相对浓度及与酶的亲和力的大小。这类抑制作用可用增加底物浓度来减弱或解除。例如,丙二酸或戊二酸的结构与琥珀酸相似,是琥珀酸脱氢酶的竞争性抑制剂。在反应中加入丙二酸或戊二酸后,就竞争性抑制了该酶的活性。当反应中增加琥珀酸的浓度后,可把与丙二酸结合的酶取代出来,减弱或解除抑制作用。竞争性抑制的效应在于增大Km, 而不改变Vmax(图3-13)。因为在I和S间竞争同一活性中心,所以需要更大的[S]才能达到 Vmax/2 ,即增高Km。当该系统被底物饱和时,Vmax不应受影响。
。因为在I和S间竞争同一活性中心,所以需要更大的[S]才能达到 Vmax/2 ,即增高Km。当该系统被底物饱和时,Vmax不应受影响。")
29
可逆抑制剂通过非共价键与酶结合 竞争性抑制剂只与游离的酶结合

30
只要底物浓度浓度足够大,酶仍然可以被底物饱和,就象没有抑制剂存在时一样,即最大反应速度应当是不变的。
竞争性抑制剂一与酶结合就阻止了底物与酶的结合,所以说底物、抑制剂与酶的结合是竞争性的。 只要底物浓度浓度足够大,酶仍然可以被底物饱和,就象没有抑制剂存在时一样,即最大反应速度应当是不变的。 因此在有竞争性抑制剂存在下, Vmax不变,但Km 增加。 吲哚是胰凝乳蛋白酶的竞争性,这是由于酶作用的底物Trp的侧链中含有吲哚基。

31
2.反竞争性抑制剂只能同ES结合 反竞争性抑制剂(uncompetitive inhibitor)仅能同ES结合,不同游离酶结合。在反竞争性抑制作用中,Vmax减小(1/Vmax增大),因为一些酶分子转变成了无活性的ESI。由于是ES复合物同I结合,Vmax的降低不能通过加入更多的底物使其发生逆转。反竞争性抑制剂也能降低Km,因为ES和ESI两者形成的平衡由于I的结合有利于向ESI复合物方向移动。代表反竞争性抑制剂浓度变化的双倒数图上的直线都有相同的斜率,这是Km和Vmax按比例减小的一种图像特征(图3-14)。这类抑制作用通常仅出现在多底物反应中。反竞争性抑制剂不需要与底物相似。
仅能同ES结合,不同游离酶结合。在反竞争性抑制作用中,Vmax减小(1/Vmax增大),因为一些酶分子转变成了无活性的ESI。由于是ES复合物同I结合,Vmax的降低不能通过加入更多的底物使其发生逆转。反竞争性抑制剂也能降低Km,因为ES和ESI两者形成的平衡由于I的结合有利于向ESI复合物方向移动。代表反竞争性抑制剂浓度变化的双倒数图上的直线都有相同的斜率,这是Km和Vmax按比例减小的一种图像特征(图3-14)。这类抑制作用通常仅出现在多底物反应中。反竞争性抑制剂不需要与底物相似。")
32
反竞争性抑制剂只与ES结合 反竞争性抑制剂只与ES结合,而不与游离酶结合。

33
在反竞争性抑制作用中,Vmax减小(1/Vmax增大),Km减小(即1/Km的绝对值增大)。
,Km减小(即1/Km的绝对值增大)。")
34
3.非竞争性抑制剂既能同酶结合又能同ES结合
酶可同时与底物和抑制剂结合,两者无竞争作用,故称为非竞争性抑制作用(noncompetitive inhibition )。酶与底物结合后也可和抑制剂结合:ES + I - ESI;酶与抑制剂结合后也可和底物结合:EI + S - ESI;可见:底物和抑制剂与酶的结合部位不同。但酶-底物-抑制剂复合物不能再转变成产物,故酶的活性下降。 这类抑制剂与酶的活性中心以外的基团结合,其结构与底物无类似之处,其抑制作用不能用提高底物浓度来解除。其抑制效应在于降低Vmax而不改变Km(图3-15)。
。酶与底物结合后也可和抑制剂结合:ES + I - ESI;酶与抑制剂结合后也可和底物结合:EI + S - ESI;可见:底物和抑制剂与酶的结合部位不同。但酶-底物-抑制剂复合物不能再转变成产物,故酶的活性下降。 这类抑制剂与酶的活性中心以外的基团结合,其结构与底物无类似之处,其抑制作用不能用提高底物浓度来解除。其抑制效应在于降低Vmax而不改变Km(图3-15)。")
35
非竞争性抑制剂与ES和E都结合 非竞争性抑制剂与底物结合部位不同,所以他既可与E结合,也可与ES结合,生成的EI和ESI都是失活形式的复合物。

36
在非竞争性抑制作用中, Km不变, Vmax变小。

37
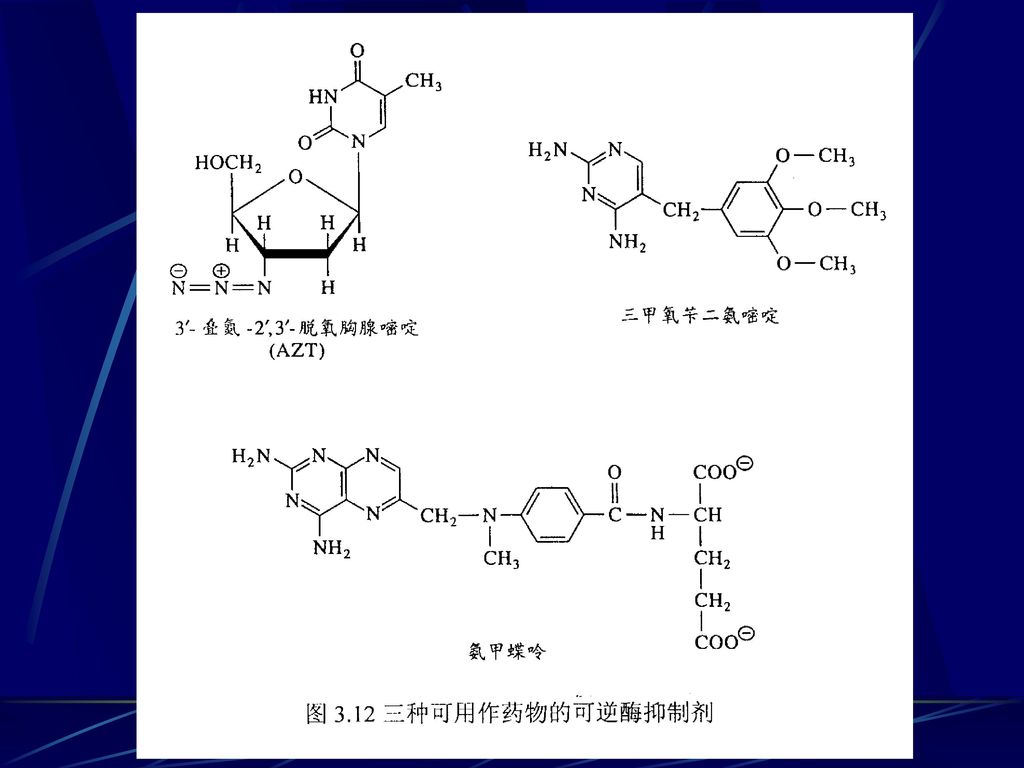
可逆的酶抑制剂常用于酶学研究和临床 临床上通过改变酶活性可以提供非常有效的治疗手段。在癌症和微生物或病毒感染中,药物治疗的目的是在不伤害宿主细胞的条件下防止受侵害细胞或病毒的繁殖。例如通过抑制病毒DNA合成所需要的酶就可使病毒感染减轻。 核苷3ˊ-叠氮-2ˊ,3ˊ-脱氧胸腺嘧啶(AZT,)是第一个用于治疗获得性免疫缺陷综合症(AIDS)的药物,它通过代谢可转换为相应的AZT 5ˊ-三磷酸,该三磷酸化合物抑制催化由病毒RNA模板合成DNA的逆转录酶。
是第一个用于治疗获得性免疫缺陷综合症(AIDS)的药物,它通过代谢可转换为相应的AZT 5ˊ-三磷酸,该三磷酸化合物抑制催化由病毒RNA模板合成DNA的逆转录酶。")
38
氨甲蝶呤和三甲氧苄二氨嘧啶都是二氢叶酸还原酶的竞争性抑制剂,该酶是胸腺嘧啶单磷酸(TMP)合成所必需的酶。
氨甲蝶呤可以抑制脊椎动物二氢叶酸还原酶和被用于选择性杀伤快速分裂的细胞,是最成功的癌症化疗制剂之一。 三甲氧苄二氨嘧啶是原核生物二氢叶酸还原酶的一个有潜力的选择性抑制剂,但对人的还原酶的抑制很弱,所以常被用于某些细菌感染和疟疾的治疗。

40
另外,磺胺药是以对氨基苯磺酰胺为主要成分的消炎药(杀菌药),它的结构非常类似于细菌生长繁殖所需要的叶酸中对氨基苯甲酸,叶酸在二氢叶酸合成酶的催化下生成二氢叶酸,所以磺胺药与叶酸竞争结合二氢叶酸合成酶,因此磺胺药是细菌二氢叶酸合成酶的竞争性抑制剂。 人由于可以直接从食物中获得叶酸,所以基本上不受磺胺药的作用。但细菌不能利用外源的叶酸。

41
磺胺药中的主要成分 叶 酸

42
Section 5 酶 作 用 的 机 制 两个基本的﹑且相互关联的原理为酶的作用机制提供了普遍性的解释:
●酶的催化效率最终源自发生在酶和它的底物之间的多种弱的作用的形成和相互作用所释放的自由能。这种结合能既贡献于酶作用的专一性,又贡献于它的催化作用; ●在反应的转换态中使这样的弱的相互作用处于最佳状态。 酶的活性部位本身与底物是互不相补的,但对反应转换态来说则是互补的,是可以被催化的.

43
一.酶和底物间的相互作用力是催化作用的关键
1.底物与酶的邻近极大地有利于酶促反应 化学反应的速度取决于反应物分子的有效碰撞。处在溶液中的反应物分子只有通过彼此扩散相遇才有可能发生反应。在生理条件下,一个典型的溶质分子和酶分子活性部位的结合的频率大约是108~109 mol·L-1S-1。如果反应物之间有静电吸引,相遇的频率可能是很高的.底物同酶的结合是一种非常快速的二级反应.如果因酶的存在能加快底物分子的扩散相遇的频率,酶促反应速度就能加快.1975年,W. Jencks提出了魔力效应(circe effect)解释酶活性部位对底物分子的强烈吸引力.目前对这种效应的证实尚缺乏证据,但认为可能涉及两者间的静电吸引力。
解释酶活性部位对底物分子的强烈吸引力.目前对这种效应的证实尚缺乏证据,但认为可能涉及两者间的静电吸引力。")
44
对于多底物酶来说,底物分子在酶活性部位的集中,使它们的有效浓度超过了它们处在溶液中的浓度.同样的道理,底物结合在催化部位的氨基酸残基附近, 增高了两个反应物的浓度.高度有效的底物浓度有利于转换态更为频繁的形成。这种现象叫做邻近效应(proximity effect) 。邻近效应需要底物分子同酶弱的结合,即这种结合是低亲和力的。因为非常紧密的结合会使催化变得无效。实验表明,邻近效应对酶促反应的贡献是使反应的效率提高104~105。

45
2.酶-底物复合物的形成伴随着熵减和去稳定 转换态出现在活化屏障峰的顶点,必须超过这一能量屏障才能进行反应。如图3-16所示,在没有催化剂存在的情况下,能障是S和S‡ (或X‡ )之间的能量差。同样地,在有酶催化的反应中(图3—16), 能障是ES和ES‡ (或EX‡ )的能量差。这种能量差越小,就越容易达到转换态,转换态的浓度越高,反应的速度就越快。酶的存在能够降低底物和它的转换态之间的能量差.如图3-16,在S转变成ES‡之前,先与酶(E)形成酶-底物复合物(ES),然后才能转变成ES‡ (或EX‡ ) ,ES‡ (或EX‡ )具有的自由能显然比S‡的低。
之间的能量差。同样地,在有酶催化的反应中(图3—16), 能障是ES和ES‡ (或EX‡ )的能量差。这种能量差越小,就越容易达到转换态,转换态的浓度越高,反应的速度就越快。酶的存在能够降低底物和它的转换态之间的能量差.如图3-16,在S转变成ES‡之前,先与酶(E)形成酶-底物复合物(ES),然后才能转变成ES‡ (或EX‡ ) ,ES‡ (或EX‡ )具有的自由能显然比S‡的低。")
46
ES与转换态(ES‡)是不同的。由于酶促反应速度由ES和ES‡之间的能量差决定,因此,这个差值越小,酶促反应速度就越快。ES复合物的能量水平的升高是通过两种途径达到的:①由S和E的结合所造成的熵减;②ES复合物由于张力、变形、去溶剂化等效应所引起的去稳定作用。熵减源于这样一个事实,即ES复合物是高度组织化的、低熵的统一体,而溶液中的E和S是处在无序的、高熵的状态。底物进入到酶的活性部位引起反应基团及其他相关基团与底物一起进入合适的位置。这种从无序到有序的转变必然导致熵的减少。 因结构张力或变形引起了ES复合物的去稳定作用是基于这样一个事实,即酶同转换态的结合比同底物的结合更紧密。当底物结合时,“契合”(fit) 的不完全性质导致底物、酶或者酶和底物变形或产生张力。这就意味着构成酶活性部位的氨基酸残基将进一步定向地使转换态的结构完善。
 的不完全性质导致底物、酶或者酶和底物变形或产生张力。这就意味着构成酶活性部位的氨基酸残基将进一步定向地使转换态的结构完善。")
47
二.酶能使转换态稳定 锁-钥模型最初提出时,是用来解释酶作用的专一性,该模型认为,酶-底物复合物是一种完全互补的形式,就象锁和钥那样。但是一种酶和它的底物是不可能完全互补的。这种完全互补会导致酶不能对底物进行催化。酶与转换态的结合是互补的,如果转换态是钥,酶就能专一地催化反应了。S、E、ES以及ES‡与催化的关系可以用假想的“磁酶”和它的底物“金属棒”之间的关系来解释(图3-17)。 ES复合物是不太稳定的,它容易解离成 E+S。这是由于前面我们已经讨论过的,即在ES复合物中,底物与酶的结合并不是处于最适状态,即不是处于完全互补的状态。换句话说,酶活性部位的基团以及能与底物发生弱的相互作用的氨基酸残基并未全部参与到同底物的结合中去。只有当ES进行转变,进入到转换态ES‡,才能使底物处于一种最适的结合状态。这就是说,酶与转换态是互补的。 为了使酶结合的底物进入到转换态,就需继续获取能量,达到转换态所必需的自由能。其中大多数所需的能量涉及到远离敏感键的部位。于是,酶和它的底物非反应部分间的相互作用提供催化反应所需的一些能量。这种能量的补偿使得反应所需的活化能进一步降低。这种仅在转换态形成的弱的相互作用是对催化作用的重要贡献。转换态的稳定对酶催化效率的贡献至少提高104~5。
。 ES复合物是不太稳定的,它容易解离成 E+S。这是由于前面我们已经讨论过的,即在ES复合物中,底物与酶的结合并不是处于最适状态,即不是处于完全互补的状态。换句话说,酶活性部位的基团以及能与底物发生弱的相互作用的氨基酸残基并未全部参与到同底物的结合中去。只有当ES进行转变,进入到转换态ES‡,才能使底物处于一种最适的结合状态。这就是说,酶与转换态是互补的。 为了使酶结合的底物进入到转换态,就需继续获取能量,达到转换态所必需的自由能。其中大多数所需的能量涉及到远离敏感键的部位。于是,酶和它的底物非反应部分间的相互作用提供催化反应所需的一些能量。这种能量的补偿使得反应所需的活化能进一步降低。这种仅在转换态形成的弱的相互作用是对催化作用的重要贡献。转换态的稳定对酶催化效率的贡献至少提高104~5。")
48
三、酶的酸-碱催化 几乎所有的酶促反应在某种程度上都涉及酸或碱的催化。有两种类型的酸-碱催化(Acid-base catalysis):(a)特指的酸-碱催化,是指H+和OH-对反应的加速;(b)广义的酸-碱催化,是指质子的供体(酸) 或质子的受体(碱) 对反应的加速。特指的酸-碱催化和广义的酸-碱催化在实验上是可以区别的。特指的酸-碱催化不会受缓冲液浓度的影响;而广义的酸-碱催化则因缓冲液可能供出或接受转换态的质子而影响反应速度。广义的酸-碱催化是生物化学上解释酶促反应效率的重要因素。 广义的酸催化是一种从酸转移质子的过程,能降低反应转换态的自由能。例如,非酶催化的酮式-烯醇式互变异构反应以十分缓慢的速度发生,因为它的类似负碳离子(carbanion) 的转换态具有很高的自由能(图3-18a),但是,当质子提供给氧原子时能降低转换态的负碳离子的特性,因而能加快反应(图3-18b)。 广义的碱催化是指碱对质子的吸引,同样能起到加快反应速度的作用。当然有些反应也涉及到酸-碱协同催化。 许多生物化学上的反应易遭受到广义的酸-碱催化。Asp、Glu、His、Cys、Tyr以及Lys残基的侧链在接近生理pH范围下都有它们相应的pK值,因而使得这些氨基酸残基的侧基团可以作为酸或碱催化剂。具有酸-碱催化能力的酶对pH非常敏感,因为pH影响酶活性部位的侧链质子化状态。 广义的酸或广义的碱催化可使反应速度提高10~100倍。
:(a)特指的酸-碱催化,是指H+和OH-对反应的加速;(b)广义的酸-碱催化,是指质子的供体(酸) 或质子的受体(碱) 对反应的加速。特指的酸-碱催化和广义的酸-碱催化在实验上是可以区别的。特指的酸-碱催化不会受缓冲液浓度的影响;而广义的酸-碱催化则因缓冲液可能供出或接受转换态的质子而影响反应速度。广义的酸-碱催化是生物化学上解释酶促反应效率的重要因素。 广义的酸催化是一种从酸转移质子的过程,能降低反应转换态的自由能。例如,非酶催化的酮式-烯醇式互变异构反应以十分缓慢的速度发生,因为它的类似负碳离子(carbanion) 的转换态具有很高的自由能(图3-18a),但是,当质子提供给氧原子时能降低转换态的负碳离子的特性,因而能加快反应(图3-18b)。 广义的碱催化是指碱对质子的吸引,同样能起到加快反应速度的作用。当然有些反应也涉及到酸-碱协同催化。 许多生物化学上的反应易遭受到广义的酸-碱催化。Asp、Glu、His、Cys、Tyr以及Lys残基的侧链在接近生理pH范围下都有它们相应的pK值,因而使得这些氨基酸残基的侧基团可以作为酸或碱催化剂。具有酸-碱催化能力的酶对pH非常敏感,因为pH影响酶活性部位的侧链质子化状态。 广义的酸或广义的碱催化可使反应速度提高10~100倍。")
49
四、酶的共价催化 共价催化(covalent catalysis)是指催化剂与底物之间瞬间形成共价键以提高反应速度的一种催化机制。通常,共价键的形成是通过催化剂上的亲核基团和底物的亲电基团的反应实现的。这种催化形式叫做亲核催化。 催化剂的亲核性与它的碱性密切相关。实际上,亲核催化的机制与碱催化的机制相似,所不同的只是以催化剂亲核攻击底物形成共价键代替从底物上吸引质子。生物学上重要的亲核剂是带负电荷的基团或含有容易与缺电子基团形成共价键的未共用的电子对;相反,亲电剂包括那些带正电荷的、含未饱和的价电子层的﹑或具电负性的原子。

50
共价催化的一个重要的特征是:形成的共价键越稳定,就越不容易在反应的最后一步被破坏。因此,一种好的催化剂必须兼备高亲核性和形成一种好的离去基团(leaving group)的(看似相矛盾的)性质。即是说,键形成的步骤是容易可逆的。具高度极化性(高流动性电子)的基团,例如咪唑和巯醇等功能基团具有这样的特性,因此能成为好的共价催化剂。在蛋白质分子中,这样的功能基团包括Lys的ε-氨基、His的咪唑基、Cys的巯基、 Asp的羧基和Ser的羟基。此外,一些辅酶(特别是焦磷酸硫胺素和磷酸吡哆醛)能与它们的脱辅基酶结合行使共价催化剂的功能。共价催化在酶的催化反应中占据十分突出的位置,对酶促反应速度的加速贡献大约是100倍。
的(看似相矛盾的)性质。即是说,键形成的步骤是容易可逆的。具高度极化性(高流动性电子)的基团,例如咪唑和巯醇等功能基团具有这样的特性,因此能成为好的共价催化剂。在蛋白质分子中,这样的功能基团包括Lys的ε-氨基、His的咪唑基、Cys的巯基、 Asp的羧基和Ser的羟基。此外,一些辅酶(特别是焦磷酸硫胺素和磷酸吡哆醛)能与它们的脱辅基酶结合行使共价催化剂的功能。共价催化在酶的催化反应中占据十分突出的位置,对酶促反应速度的加速贡献大约是100倍。")
51
五、丝氨酸蛋白酶的结构特点与作用机制 1.丝氨酸蛋白酶的结构特点
丝氨酸蛋白酶类包括胰蛋白酶、胰凝乳蛋白酶、弹性蛋白酶(胰肽酶E)、枯草杆菌蛋白酶(subtilisin)和其他相关酶。之所以称为丝氨酸蛋白酶,是因为它们有共同的、涉及特有的反应丝氨酸残基的作用机制;二异苯氟磷酸(DIPF)是丝氨酸蛋白酶的不可逆抑制剂,它只能与活性部位的Ser残基结合,从而导致酶的失活,这就证明了这个丝氨酸残基是酶活性所必需的。(虽然乙酰胆碱酯酶本身不是一种蛋白酶,但该酶是一个丝氨酸酯酶,并在机制上与丝氨酸蛋白酶相似)。
、枯草杆菌蛋白酶(subtilisin)和其他相关酶。之所以称为丝氨酸蛋白酶,是因为它们有共同的、涉及特有的反应丝氨酸残基的作用机制;二异苯氟磷酸(DIPF)是丝氨酸蛋白酶的不可逆抑制剂,它只能与活性部位的Ser残基结合,从而导致酶的失活,这就证明了这个丝氨酸残基是酶活性所必需的。(虽然乙酰胆碱酯酶本身不是一种蛋白酶,但该酶是一个丝氨酸酯酶,并在机制上与丝氨酸蛋白酶相似)。")
52
胰蛋白酶、胰凝乳蛋白酶和弹性蛋白酶都能完成同样的反应---肽链裂解。虽然它们的结构和作用机制是十分相似的,但是,它们却表现出很不相同的(基团)专一性。这三种酶的分子量大约是25 kD,有相似的的顺序和三维空间结构,整个分子呈椭球形。图3—19是胰蛋白酶主链在空间中的走向图解,示出了它的螺旋结构和β-片以及催化三联体(His57、Asp102和Ser195) 的位置。催化三联体在三种胰脏酶中都存在。
专一性。这三种酶的分子量大约是25 kD,有相似的的顺序和三维空间结构,整个分子呈椭球形。图3—19是胰蛋白酶主链在空间中的走向图解,示出了它的螺旋结构和β-片以及催化三联体(His57、Asp102和Ser195) 的位置。催化三联体在三种胰脏酶中都存在。")
53
2.丝氨酸蛋白酶的作用机制(以胰凝乳蛋白酶为例)
胰凝乳蛋白酶为例示于图3—20。 3.丝氨酸蛋白酶进化上的关系 胰凝乳蛋白酶、胰蛋白酶、弹性蛋白酶来自同一组织--胰脏,都是内肽酶。这三种酶被认为是由一个共同的祖先基因(ancestral gene)在进化过程中通过同源趋异进化(divergent evolution) 产生的三个基因编码的。因为: 1).这三种酶的活性中心都含有可与DIFP起反应的Ser残基; 2).在活性部位的Ser附近都含有相同的氨基酸顺序: --Gly-Asp-Ser-Gly-Gly-Pro-- 3).在它们的三级结构中都含有相同的、恒定的 Asp102-His57-Ser195- 的组合顺序(相同的电荷转换系统); 4).它们氨基酸的顺序大约有40%相同; 5) 它们有很相似的空间结构。
在进化过程中通过同源趋异进化(divergent evolution) 产生的三个基因编码的。因为: 1).这三种酶的活性中心都含有可与DIFP起反应的Ser残基; 2).在活性部位的Ser附近都含有相同的氨基酸顺序: --Gly-Asp-Ser-Gly-Gly-Pro-- 3).在它们的三级结构中都含有相同的、恒定的. Asp102-His57-Ser195- 的组合顺序(相同的电荷转换系统); 4).它们氨基酸的顺序大约有40%相同; 5) 它们有很相似的空间结构。")
54
** X-射线晶体图分析揭示了丝氨酸蛋白酶底物结合特异性
胰凝乳蛋白酶、胰蛋白酶和弹性蛋白酶结构上的微小差别反映出它们底物的特异性。胰蛋白酶与胰凝乳蛋白酶之间结构上的主要区别是胰凝乳蛋白酶的疏水性口袋的底部是一个不带电荷的氨基酸残基(图a),而胰蛋白酶是一个带电荷的天冬氨酸残基(图b),这个带负电荷的残基是造成胰蛋白酶底物特异性的原因,在ES复合物中,它与底物的带正电荷的Arg和Lys残基的侧链形成离子对。 弹性蛋白酶在三级结构上类似于胰凝乳蛋白酶,但弹性蛋白酶的结合口袋要浅得多。胰凝乳蛋白酶和胰蛋白酶结合部位的入口处有两个甘氨酸残基,而在弹性蛋白酶的入口处是比甘氨酸大得多的缬氨酸和苏氨酸残基(图c)。
,而胰蛋白酶是一个带电荷的天冬氨酸残基(图b),这个带负电荷的残基是造成胰蛋白酶底物特异性的原因,在ES复合物中,它与底物的带正电荷的Arg和Lys残基的侧链形成离子对。 弹性蛋白酶在三级结构上类似于胰凝乳蛋白酶,但弹性蛋白酶的结合口袋要浅得多。胰凝乳蛋白酶和胰蛋白酶结合部位的入口处有两个甘氨酸残基,而在弹性蛋白酶的入口处是比甘氨酸大得多的缬氨酸和苏氨酸残基(图c)。")
55
胰凝乳蛋白酶的疏水性口袋的底部是一个不带电荷的氨基酸残基

56
底物“口袋”中有一个带电荷的天冬氨酸残基

57
酶结合底物的口袋中含有缬氨酸和苏氨酸残基,限制大的底物进入

58
枯草杆菌蛋白酶和小麦胚芽丝氨酸羧肽酶Ⅱ(一种外肽酶)也是丝氨酸蛋白酶,彼此之间以及与胰凝乳蛋白酶之间在一级或三级结构上不具有共同的可辩别的关系。然而,这两种酶在它们的活性部位上具有与胰脏酶相似的催化三联体,由于这两种酶活性部位相应的催化三联体的顺序是不同的(在胰凝乳蛋白酶中是Asp102﹣His57﹣Ser195,在枯草杆菌蛋白酶中是Asp32﹣His64﹣Ser221,在小麦胚芽丝氨酸羧肽酶Ⅱ中是Asp338﹣His397-Ser146),因此,它们从一个共同的祖先蛋白(ancestor protein)进化而来是很不可能的。这些酶显然构成了一个异源趋同进化(convergent evolution)的例子。
也是丝氨酸蛋白酶,彼此之间以及与胰凝乳蛋白酶之间在一级或三级结构上不具有共同的可辩别的关系。然而,这两种酶在它们的活性部位上具有与胰脏酶相似的催化三联体,由于这两种酶活性部位相应的催化三联体的顺序是不同的(在胰凝乳蛋白酶中是Asp102﹣His57﹣Ser195,在枯草杆菌蛋白酶中是Asp32﹣His64﹣Ser221,在小麦胚芽丝氨酸羧肽酶Ⅱ中是Asp338﹣His397-Ser146),因此,它们从一个共同的祖先蛋白(ancestor protein)进化而来是很不可能的。这些酶显然构成了一个异源趋同进化(convergent evolution)的例子。")
59
Section 酶活性的调节 酶活性的调节是酶作为生物催化剂区别于非生物催化剂的一个重要的标志,也是生物体内物质代谢的重要调节方式。酶活性的调节大致包括酶原的激活、同工酶调节、别构调节和共价修饰调节。酶的一些特殊组织结构形式对酶活性的控制也是重要的。 一.酶原的激活(以胰凝乳蛋白酶原的激活为例) 从无活性的蛋白质原或酶原转变成有活性的蛋白质或酶的过 程即称激活或活化。胰凝乳蛋白酶原激活过程如图3-21所示。 激活的关键是选择性地切断了Arg15和Ile16之间的肽键,使Ile16游离。在中性条件下,Ile16的α﹣NH3+能和Asp194的β﹣COO-通过静电作用形成离子键,有助于Ser195和His57之间以及His57和 Asp102之间通过氢键形成一个“电荷转接系统”,使Ser195的侧链具有更强的亲核性,有利于和底物进行作用,于是就表现出胰凝乳蛋白酶的完整的催化功能。 酶原的激活是生物体的一种调控方式,具有重要的生理意义。
 从无活性的蛋白质原或酶原转变成有活性的蛋白质或酶的过. 程即称激活或活化。胰凝乳蛋白酶原激活过程如图3-21所示。 激活的关键是选择性地切断了Arg15和Ile16之间的肽键,使Ile16游离。在中性条件下,Ile16的α﹣NH3+能和Asp194的β﹣COO-通过静电作用形成离子键,有助于Ser195和His57之间以及His57和 Asp102之间通过氢键形成一个 电荷转接系统 ,使Ser195的侧链具有更强的亲核性,有利于和底物进行作用,于是就表现出胰凝乳蛋白酶的完整的催化功能。 酶原的激活是生物体的一种调控方式,具有重要的生理意义。")
60
二、同工酶 来自同一生物不同组织或同一细胞的不同亚细胞结构﹑能催化相同反应的酶叫做同工酶(isozyme)。同工酶是多亚基蛋白,通常由四个以上的亚基构成。同一种酶的不同的同工酶的亚基通常只有一个或少数氨基酸残基的差别,但也可能是不同的蛋白质。同工酶在化学上是可以区分的,可以通过离子交换柱层析法和电泳法进行鉴定。来自哺乳动物的乳酸脱氢酶(lactate dehydrogenase,LDH) 是一种四聚体蛋白,从不同组织提取物中可以区分出五种不同的形式,并鉴定出两种不同的亚基,即心肌型的H(或A)亚基以及骨骼肌型的M(或B) 亚基。LDH的五种同工酶都是由这两种亚基以不同比例装配而成的四聚体;H4、H3M、H2M2、HM3和M4(图3—22) 。 同一种酶的不同的同工酶在对底物的亲和力和Vmax以及对产物抑制的敏感性等通常都是不同的,这反映出代谢上的需要。谷氨酰胺合成酶(glutamine synthetase, GS)是氮素代谢中的一种重要的酶,该酶催化依赖于ATP、由谷氨酸和NH4+合成谷氨酰胺的反应。在高等植物的叶片中,大多存在两种不同类型的GS同工酶,即胞质型的GS1和叶绿体型的GS2。这两种同工酶在发育上以及在代谢中起着非常重要的作用。
。同工酶是多亚基蛋白,通常由四个以上的亚基构成。同一种酶的不同的同工酶的亚基通常只有一个或少数氨基酸残基的差别,但也可能是不同的蛋白质。同工酶在化学上是可以区分的,可以通过离子交换柱层析法和电泳法进行鉴定。来自哺乳动物的乳酸脱氢酶(lactate dehydrogenase,LDH) 是一种四聚体蛋白,从不同组织提取物中可以区分出五种不同的形式,并鉴定出两种不同的亚基,即心肌型的H(或A)亚基以及骨骼肌型的M(或B) 亚基。LDH的五种同工酶都是由这两种亚基以不同比例装配而成的四聚体;H4、H3M、H2M2、HM3和M4(图3—22) 。 同一种酶的不同的同工酶在对底物的亲和力和Vmax以及对产物抑制的敏感性等通常都是不同的,这反映出代谢上的需要。谷氨酰胺合成酶(glutamine synthetase, GS)是氮素代谢中的一种重要的酶,该酶催化依赖于ATP、由谷氨酸和NH4+合成谷氨酰胺的反应。在高等植物的叶片中,大多存在两种不同类型的GS同工酶,即胞质型的GS1和叶绿体型的GS2。这两种同工酶在发育上以及在代谢中起着非常重要的作用。")
61
三、多酶复合物和多功能酶 在某些情况下,催化同一代谢反应顺序的几种酶彼此组合在一起,构成了一种多酶复合物(multienzyme complex)。这种多酶复合物的存在有其显著优点。多酶复合物为代谢物的流动开辟了一个通道。这样的通道允许一个反应的产物直接转移到下一个酶的活性部位,不进入到溶剂中,减少代谢中间物达到下一个酶的转移时间,并增高中间物的局部浓度而使反应加速。丙酮酸脱氢酶复合物是由三种不同的酶构成的一种多酶复合物,该复合物催化丙酮酸转变成乙酰CoA,后者进入柠檬酸循环。丙酮酸脱氢酶复合物是多酶复合物的一个典型例子。 有些多酶复合物与膜结合在一起,在膜结构中有着相应的定位关系,例如催化电子传递反应的呼吸链,是由多个酶复合物组成,这些酶复合物在线粒体内膜上有着严格的定位关系,为催化电子的传递和跨膜的电化学梯度的产生提供了结构基础。
。这种多酶复合物的存在有其显著优点。多酶复合物为代谢物的流动开辟了一个通道。这样的通道允许一个反应的产物直接转移到下一个酶的活性部位,不进入到溶剂中,减少代谢中间物达到下一个酶的转移时间,并增高中间物的局部浓度而使反应加速。丙酮酸脱氢酶复合物是由三种不同的酶构成的一种多酶复合物,该复合物催化丙酮酸转变成乙酰CoA,后者进入柠檬酸循环。丙酮酸脱氢酶复合物是多酶复合物的一个典型例子。 有些多酶复合物与膜结合在一起,在膜结构中有着相应的定位关系,例如催化电子传递反应的呼吸链,是由多个酶复合物组成,这些酶复合物在线粒体内膜上有着严格的定位关系,为催化电子的传递和跨膜的电化学梯度的产生提供了结构基础。")
62
多功能酶(multifunctional enzyme)是酶的另一种有效的组织结构形式。多功能酶或是由单一的、具多个活性部位的多肽链构成,或是由两个、各具多个活性部位的多肽链组成。几个相关的酶活性共价结合在同一个蛋白质分子中是基因在进化中融合的结果。例如,在细菌和植物中,催化脂肪酸生物合成的酶是以离散的形式分布在细胞质中,而在真菌中,这些酶通过编码它们的基因的融合,分别融合成两个不同的多肽链(即α和β亚基),在哺乳动物中,则组合成单一的、多功能的、具所有酶活性的多肽链。除具有多酶复合物所具有的优点外,多功能酶最突出的优点是它们的合成是协调一致的,而且一种共价结合在一起的多功能复合物很可能比由非共价相互作用所形成的复合物更稳定。
是酶的另一种有效的组织结构形式。多功能酶或是由单一的、具多个活性部位的多肽链构成,或是由两个、各具多个活性部位的多肽链组成。几个相关的酶活性共价结合在同一个蛋白质分子中是基因在进化中融合的结果。例如,在细菌和植物中,催化脂肪酸生物合成的酶是以离散的形式分布在细胞质中,而在真菌中,这些酶通过编码它们的基因的融合,分别融合成两个不同的多肽链(即α和β亚基),在哺乳动物中,则组合成单一的、多功能的、具所有酶活性的多肽链。除具有多酶复合物所具有的优点外,多功能酶最突出的优点是它们的合成是协调一致的,而且一种共价结合在一起的多功能复合物很可能比由非共价相互作用所形成的复合物更稳定。")
63
四. 别构酶 一个多酶体系催化的总速度取决于其中反应速度最慢的一步反应,该反应限制着整个反应链的反应速度,故称“限速反应” 或“限速步骤” 。多数具有自我调节能力的多酶体系的第一步反应就是“限速反应”,催化限速反应的酶就是一种调节酶。调节酶是一类双功能的酶,这类酶具有催化功能,也具有同调节物分子结合而改变酶反应速度的功能。这类调节酶通常是别构酶。

64
1.别构酶(或变构酶)(allosteric enzyme)结构特点
●为寡聚体酶,即多体酶,含二个或多个亚基,比一般单体酶要大得多和复杂得多。 ●含有活性部位和调节(别构)部位,活性部位负责对底物的结合与催化,调节部位负责调节酶催化反应的速度。 ●每一种别构酶的调节部位只能与它的专一性调节物或效应物分子非共价地结合。 2.别构效应 当专一性的调节物结合到调节部位时,酶的催化活性发生改变,这种改变是由于调节物与调节部位结合后诱导酶分子的构象发生变化,使酶的活性部位对底物的结合与催化作用受到影响,从而调节酶促反应速度及代谢过程。此效应称别构效应或变构效应(allosteric effect)。
部位,活性部位负责对底物的结合与催化,调节部位负责调节酶催化反应的速度。 ●每一种别构酶的调节部位只能与它的专一性调节物或效应物分子非共价地结合。 2.别构效应. 当专一性的调节物结合到调节部位时,酶的催化活性发生改变,这种改变是由于调节物与调节部位结合后诱导酶分子的构象发生变化,使酶的活性部位对底物的结合与催化作用受到影响,从而调节酶促反应速度及代谢过程。此效应称别构效应或变构效应(allosteric effect)。")
65
3.调节物或效应物 ★负调节效应物 负调节物分子是该酶底物以外的分子,往往是多酶体系的末端产物(即反应链中最后一个酶催化的产物), 末端产物积累超出细胞需要,对该反应体系中催化限速反应的酶造成抑制(该酶往往就是别构酶),从而终止该产物的合成。这种抑制作用称为反馈抑制(feedback inhibition)(图3-23)。 ★正调节效应物 有的别构酶可被它的专一性调节物激活,这种调节物称作正调节物或正效应物。正调节物在很多情况下是酶的底物分子本身。此类型的别构酶又称作底物调节酶(homotropic enzyme)或同位(促)酶,因底物和调节物是同一的。这类别构酶与底物的过量积累而必须通过后续反应移走的情况有关。 ▼别构酶受底物分子调节的效应称作同位(促)效应(homotropic effect); ▼别构酶受底物以外分子(例如末端产物)调节的效应称作异位(促)效应,此类别构酶亦称异位酶(heterotropic enzyme)。异位(促)效应有二种情况:异位抑制和异位激活。
, 末端产物积累超出细胞需要,对该反应体系中催化限速反应的酶造成抑制(该酶往往就是别构酶),从而终止该产物的合成。这种抑制作用称为反馈抑制(feedback inhibition)(图3-23)。 ★正调节效应物 有的别构酶可被它的专一性调节物激活,这种调节物称作正调节物或正效应物。正调节物在很多情况下是酶的底物分子本身。此类型的别构酶又称作底物调节酶(homotropic enzyme)或同位(促)酶,因底物和调节物是同一的。这类别构酶与底物的过量积累而必须通过后续反应移走的情况有关。 ▼别构酶受底物分子调节的效应称作同位(促)效应(homotropic effect); ▼别构酶受底物以外分子(例如末端产物)调节的效应称作异位(促)效应,此类别构酶亦称异位酶(heterotropic enzyme)。异位(促)效应有二种情况:异位抑制和异位激活。")
66
调节酶一般都是寡聚体 上面是一个多酶催化途径,总的结果是由A生成P。终产物是调节酶E1的一个别构抑制剂,E1催化第一个关键的反应。
这种类型的调节称为反馈抑制。一个调节酶有一个位于它的催化中心以外的第二个配体结合部位,这个部位称为调节部位。酶构象的变化是与一个别构激活剂或抑制剂和酶的调节部位的结合有关,这种构象的变化传递给酶的活性部位,活性部位形状的变化改变了酶的活性。

67
4.别构酶的动力学特征 别构酶不服从底物浓度对酶反应速度影响的米氏方程,(即不是双曲线而是S型曲线)(图3-24),调节物分子所造成的抑制作用也不服从典型的竞争性或非竞争性抑制作用的数量关系,也不能用Km来表示最大半反应速度(Vmax/2)所对应的底物浓度。最大半反应速度所对应的底物浓度用K0.5表示(或表现Km)。 细菌天冬氨酸转氨甲酶(ATCase)是研究很详细的一种酶。 5.别构酶亚基之间的通讯关系 有二种模型可解释别构酶亚基间的通讯联系:序变模型(KNF型)和齐变模型(MWC模型)可以用来解释亚基间的通讯联系(图3-25)。
(图3-24),调节物分子所造成的抑制作用也不服从典型的竞争性或非竞争性抑制作用的数量关系,也不能用Km来表示最大半反应速度(Vmax/2)所对应的底物浓度。最大半反应速度所对应的底物浓度用K0.5表示(或表现Km)。 细菌天冬氨酸转氨甲酶(ATCase)是研究很详细的一种酶。 5.别构酶亚基之间的通讯关系. 有二种模型可解释别构酶亚基间的通讯联系:序变模型(KNF型)和齐变模型(MWC模型)可以用来解释亚基间的通讯联系(图3-25)。")
68
五、共价调节酶 共价调节酶是另外一类重要的调节酶,其活性是通过酶分子的共价修而调节的。在这类酶中,重要的例子是肌肉和肝脏中的糖原磷酸化酶。该酶催化如下反应: (Glucose)n + Pi →(Glucose)n-1 + Glc-1-P 糖原磷酸化酶以二种形式出现:有活性的磷酸化酶a, 低活性的磷酸化酶b(图3-26)。糖原磷酸化酶a和b皆由二个相同的亚基构成的二聚体;其结构差别只是磷酸化和去磷酸化。
。糖原磷酸化酶a和b皆由二个相同的亚基构成的二聚体;其结构差别只是磷酸化和去磷酸化。")
69
作业题: 1. 不同pH研究表明,某酶有二个残基对酶的催化反应是很重要,它们的pKa分别为≈4和≈10,化学修饰实验表明,Glu和Lys是该酶活性所必需的,该酶的最大催化活性的pH近中性,请把这两个残基和它们的pKa值联系起来解释它们在酶促反应中的作用。 2. 某种酶的共价催化机制取决于它的活性部位的半胱氨酸残基存在,该残基侧链的pKa值是8,它附近残基的突变改变了酶活性中心的微环境,结果使它的pKa值增至10,这种突变引起酶反应速度增高还是降低?为什么? 3. 为什么 RNaseA 不能催化DNA的水解? 4. 为什么胰凝乳蛋白酶不能象胰蛋白酶那样自我激活? 5. 酶A和酶B催化相同的反应,并可得到如图所示的反应曲线, 哪种酶在低[S]比较有效? 哪种酶在高[S]比较有效?

70
图3-1 返回

71
图3-2 返回

72
图3-3 返回

73
图3-4 返回

74
图3-6 返回

75
图3-7 返回 (1). (2)
. (2)")
76
图3-8 返回

77
图3-9 返回

78
图3-10 返回

79
a b 图3-11 c 返回

80
图3-12 返回

81
图3-13 返回

82
图3-14 返回

83
图3-15 返回

84
图3-16 返回

85
图3-17 返回

86
图3-18 返回

87
图3-19 返回

88
图3-20 返回

89
图3-21 返回

90
图3-22 返回

91
图3-24 返回

92
图3-25 返回

93
图3-26 返回

94
图3-8a 返回

95
图3-23 返回



:核蛋白体组成成分 转移 RNA ( tRNA ):转运氨基酸 信使 RNA ( mRNA ):蛋白质合成模板 不均一核 RNA ( hnRNA ):成熟 mRNA 的前体 小核 RNA ( snRNA ):>")






 2004.1 酶.>")

蛋白质的降解: 外源蛋白的消化 内源性蛋白的选择性降解 (2)氨基酸的分解代谢:>")

.>")


 主要内容: 酶催化反应速率与酶活的测定 底物浓度对酶促反应速度的影响>")



