Download presentation
Presentation is loading. Please wait.

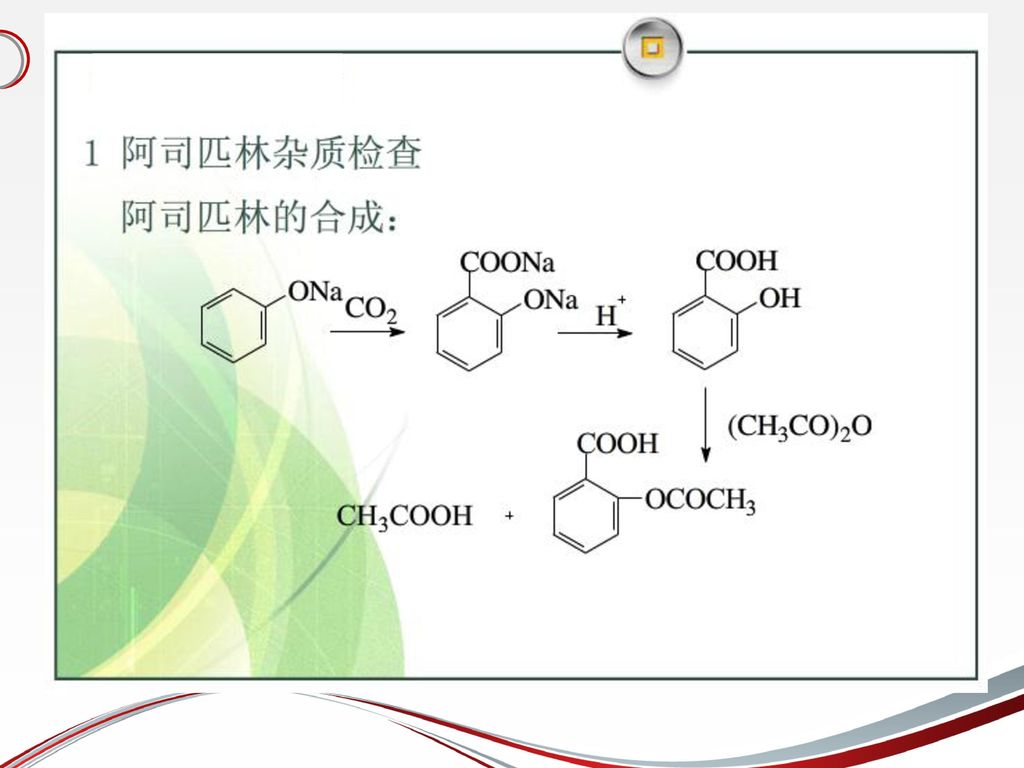
1
第四节 原料药及制剂检查

2
第一节 质量标准 第二节 原料药理化性质 第三节 鉴别
第一节 质量标准 第二节 原料药理化性质 第三节 鉴别 前面我们介绍了药品标准的制定,理化性质和鉴别的方法,这节主要是介绍原料和制剂的检测,因为制剂包含有很多辅料,附着剂等,所以对于其的检测和原料药会有所不同,所以会分别对其进行介绍

3
一、原料药检查 《中国药典》(2005年版)凡例中规定:“项下包括安全性、 有效性、均一性和纯度要求四个方面”。 药品的安全性:热原检查、毒性试验、刺激性试验、过敏 试验、升压或降压物质检查等内容。 药品的有效性:是以动物试验为基础,最终以临床疗效来 评价的。 药品的均一性:主要指制剂含量的均匀性,溶出度或释放 度的均一性,装量差异及生物利用度的均一性。 药品的纯度:主要是指对各类杂质的检查及主药的含量测 定。 热原检测,指检测该药物是否能引起恒温动物体温异常升高。由于家兔对热原的反应与人基本相似,家兔法为各国药典规定的检查热原的法定方法。除此以外,某些细菌的代谢产物、细菌尸体及内毒素也会产生发热现象,所以部分药品还要进行内毒素检测,细菌内毒素检查法系利用鲎试剂来检测或量化由革兰阴性菌产生的细菌内毒素,以判断供试品中细菌内毒素的限量是否符合规定的一种方法. 毒性试验在药品注册中已有明确的说明。毒性试验分急性毒性、亚慢性毒性和慢性毒性试验,也包括特殊毒性试验,如致畸、致癌试验、免疫毒性、遗传毒性及神经毒性试验。 刺激性试验 主要用于考察药品对组织是否引起红肿、充血、变性、坏死等刺激症状,以判定制剂的局部毒性大小,为选择合理给药方法提供参考,或采用适当措施减少或消除其刺激。 过敏试验 很多人应该都做过,如青霉素皮试,破伤风针的皮试,这些都是过敏性试验 升压或降压物质检查,一般升压药物检测是用雄性大鼠,而降压物质检测试验哦那个雌性的猫进行。 对于药品的安全性,将在后面为大家系统的介绍。
凡例中规定: 项下包括安全性、 有效性、均一性和纯度要求四个方面 。 药品的安全性:热原检查、毒性试验、刺激性试验、过敏 试验、升压或降压物质检查等内容。 药品的有效性:是以动物试验为基础,最终以临床疗效来 评价的。 药品的均一性:主要指制剂含量的均匀性,溶出度或释放 度的均一性,装量差异及生物利用度的均一性。 药品的纯度:主要是指对各类杂质的检查及主药的含量测 定。 热原检测,指检测该药物是否能引起恒温动物体温异常升高。由于家兔对热原的反应与人基本相似,家兔法为各国药典规定的检查热原的法定方法。除此以外,某些细菌的代谢产物、细菌尸体及内毒素也会产生发热现象,所以部分药品还要进行内毒素检测,细菌内毒素检查法系利用鲎试剂来检测或量化由革兰阴性菌产生的细菌内毒素,以判断供试品中细菌内毒素的限量是否符合规定的一种方法. 毒性试验在药品注册中已有明确的说明。毒性试验分急性毒性、亚慢性毒性和慢性毒性试验,也包括特殊毒性试验,如致畸、致癌试验、免疫毒性、遗传毒性及神经毒性试验。 刺激性试验 主要用于考察药品对组织是否引起红肿、充血、变性、坏死等刺激症状,以判定制剂的局部毒性大小,为选择合理给药方法提供参考,或采用适当措施减少或消除其刺激。 过敏试验 很多人应该都做过,如青霉素皮试,破伤风针的皮试,这些都是过敏性试验. 升压或降压物质检查,一般升压药物检测是用雄性大鼠,而降压物质检测试验哦那个雌性的猫进行。 对于药品的安全性,将在后面为大家系统的介绍。")
4
(一)杂质检查的内容 1.一般杂质的检查 一般杂质的检查是指对氯化物、硫酸盐、铁 盐、砷盐、铵盐、重金属、酸碱度、溶液颜色、 澄清度、水分、干燥失重、炽灼残渣、易碳化物 、有机溶剂残留物等项目的检查。 一般杂质的检查是指对氯化物、硫酸盐、铁盐、砷盐、铵盐、重金属、酸碱度、溶液颜色、澄清度、水分、干燥失重、炽灼残渣、易碳化物、有机溶剂残留物等项目的检查。

5
2.特殊杂质的检查 相对于一般杂质而言,特殊杂质是指在某药 的生产和贮藏过程中,有可能引入的仅属某药特 有的一些杂质,其中有些杂质的化学结构明确并 有其标准品或对照品。

8
螺内酯中要求检查巯基化合物,方法为碘量法; 糊精中要求检查还原糖,方法为重量法; 醋酸曲安奈德中要求检查硒,方法为氧瓶燃烧-UV 法。
需要进行特殊杂质检查的例子还有: 螺内酯中要求检查巯基化合物,方法为碘量法; 糊精中要求检查还原糖,方法为重量法; 醋酸曲安奈德中要求检查硒,方法为氧瓶燃烧-UV 法。 碘量法是氧化还原滴定法中,应用比较广泛的一种方法。 重量法就是根据单质或化合物的重量,计算出在供试品中的含量的定量方法 本法系将有机药物在充满氧气的燃烧瓶中进行燃烧,待燃烧产物被吸入吸收液后,再采用适宜的分析方法来检查或测定卤素或硫等元素的含量。 精密称取供试品(如为固体,应研细),置于无灰滤纸中心,按虚线折叠后,固定于铂丝下端的网内或螺旋处,使尾部露出。如为液体供试品,可在透明胶纸和滤纸做成的纸袋中称样。另在燃烧瓶内加入硝酸溶液(1→30)25ml为吸收液,并将瓶口用水湿润,小心急速通入氧气约1分钟(通气管应接近液面,使瓶内空气排尽),立即用表面皿覆盖瓶口,移置他处;点燃包有供试品的滤纸尾部,迅速放入燃烧瓶中,按紧瓶塞,用水少量封闭瓶口,俟燃烧完毕(应无黑色碎片),充分振摇,使生成的烟雾完全吸入吸收液中,放置15分钟,用15ml水分次冲洗瓶塞及铂丝,合并洗液及吸收液。 将上述硒对照溶液与供试品溶液分别用氨试液调节pH至2.0±0.2后,转移至分液漏斗中,用水少量分次洗涤烧杯,洗液并入分液漏斗中,使成60ml,各加盐酸羟胺溶液(1→2)1ml,摇匀后,立即精密加二氨基萘试液5ml,摇匀,在室温下放置100分钟,精密加环己烷5ml,强烈振摇2分钟,静置分层,弃去水层,环己烷层用无水硫酸钠脱水后,照分光光度法,在378nm的波长处分别测定吸收度。供试品溶液的吸收度不得大于硒对照溶液的吸收度。
,置于无灰滤纸中心,按虚线折叠后,固定于铂丝下端的网内或螺旋处,使尾部露出。如为液体供试品,可在透明胶纸和滤纸做成的纸袋中称样。另在燃烧瓶内加入硝酸溶液(1→30)25ml为吸收液,并将瓶口用水湿润,小心急速通入氧气约1分钟(通气管应接近液面,使瓶内空气排尽),立即用表面皿覆盖瓶口,移置他处;点燃包有供试品的滤纸尾部,迅速放入燃烧瓶中,按紧瓶塞,用水少量封闭瓶口,俟燃烧完毕(应无黑色碎片),充分振摇,使生成的烟雾完全吸入吸收液中,放置15分钟,用15ml水分次冲洗瓶塞及铂丝,合并洗液及吸收液。 将上述硒对照溶液与供试品溶液分别用氨试液调节pH至2.0±0.2后,转移至分液漏斗中,用水少量分次洗涤烧杯,洗液并入分液漏斗中,使成60ml,各加盐酸羟胺溶液(1→2)1ml,摇匀后,立即精密加二氨基萘试液5ml,摇匀,在室温下放置100分钟,精密加环己烷5ml,强烈振摇2分钟,静置分层,弃去水层,环己烷层用无水硫酸钠脱水后,照分光光度法,在378nm的波长处分别测定吸收度。供试品溶液的吸收度不得大于硒对照溶液的吸收度。")
9
有关物质属于特殊杂质 类。通常是在某药的生 产和贮存过程中,有可 能引入的特殊杂质。而 这些特殊杂质的化学结 构往往与主药相似,但 不甚明确或虽结构明确 ,但难以获得标准品。
例如,托吡卡胺、过氧苯甲酰、华法林钠等药物均用TLC法作为分离方法,以自身对照法进行有关物质的杂质限量检查。

10
按照我国新药报批的要求,在新药的研究中,要尽可能搞清楚有关物质的化学结构,必要时要做其药理、毒理试验;对有关物质的检查方法,应首选色谱法,

11
(二)杂质检查方法的基本要求 色谱条件 考察色谱法 的有效性
对杂质检查的基本要求是:要研究方法的基本原理、 专属性、灵敏性、试验条件的最佳化。 对于色谱法,还要研究其分离能力。 比如,用该药的粗制品,或用成品加中间体的混合物,或将成品用强酸、强碱、光照、加热进行处理,然后,在既定的色谱条件下进行样品的分离,以考察色谱法的有效性。 色谱条件 考察色谱法 的有效性

12
(三)确定杂质检查及其限量的基本原则 1. 针对性 如果研究的药物属新药,应按照新药报批的要求逐 项进行研究并将试验结果整理成报批资料。
对一般杂质的检查,针对剂型及生产工艺,应尽可能考察 有关项目。 对特殊杂质或有关物质的研究,也应针对工艺及贮藏过程 确定待检查杂质的数量及其限度。 对毒性较大的杂质如砷、氰化物等亦应严格控制。

13
2.合理性 在新药质量标准的研究阶段,检查的项目应尽可能全 面考察,但在制订该药质量标准时应合理确定其检查 的项目。
在新药质量标准的研究阶段,检查的项目应尽可能全面考察,但在制订该药质量标准时应合理确定其检查的项目。比如:对于砷,在研究阶段,肯定要进行检查。但实际上许多药的检查项下并没有砷的检查。根本原因是该药不含砷,或含量极低(如小于百万分之一)。对于此种药物,砷的检查项不列入质量标准更为合理。
。对于此种药物,砷的检查项不列入质量标准更为合理。")
14
安全有效 应根据新药报批的要求,根据生产工艺水平、参考有 关文献及各国药典,综合考虑确定一个比较合理的标 准。
因此,对杂质限度的确定是很重要的。应从安全有效的角度出发,标准太低不行,标准太高,生产上难以达到也不行。总之,应根据新药报批的要求,根据生产工艺水平、参考有关文献及各国药典,综合考虑确定一个比较合理的标准。 应根据新药报批的要求,根据生产工艺水平、参考有 关文献及各国药典,综合考虑确定一个比较合理的标 准。

15
二、制剂检查 各种制剂的检查项目,除应符合相应的制剂通则中的 共性规定外,还应根据其特性、工艺及稳定性考察, 制订其他项目。
各种制剂的检查项目,除应符合相应的制剂通则中的共性规定外,还应根据其特性、工艺及稳定性考察,制订其他项目,如口服固体制剂(以片剂、胶囊剂为主)应制订含量均匀度,溶出度、释放度,有关物质(或已知杂质)等检查。注射剂应制订pH值、颜色(或溶液的颜色),有关物质(或已知杂质)、注射用粉剂或冻干品的干燥失重或水分、大输液的重金属与不溶性微粒等检查。
应制订含量均匀度,溶出度、释放度,有关物质(或已知杂质)等检查。注射剂应制订pH值、颜色(或溶液的颜色),有关物质(或已知杂质)、注射用粉剂或冻干品的干燥失重或水分、大输液的重金属与不溶性微粒等检查。")
16
(一)含量均匀度 含量均匀度系指小剂量片剂、膜剂、胶囊剂或注射用无菌粉 剂等制剂中的每片(个)含量偏离标示量的程度。
《中国药典》对含量均匀度应用的指导原则是: 1. 主要适用于规格含量小于10 mg(含10 mg)的品种 2. 用于单个制剂(片,个或支)主药含量少,辅料较 多,目标难混匀(主药含量在5%以下)的品种; 3. 用于急救、毒剧药品或安全范围小的品种; 4. 主要适用于口服固体制剂的品种。 含量均匀度是考察制剂工艺水平的重要指标之一。以前是用装(重)量差异来衡量的。但随着科学技术的发展及临床要求,装(重)量差异检查不能满足对小剂量制剂质量控制的要求。重(装)量差异是检查主药和辅料的总限度,没有明确考察主药的均一性,而主药是起治疗作用的,尤其是对那些治疗剂量与中剂量比较接近的品种,控制其含量均匀度显得尤为重要。 除另有规定外,测定时取样数量和含量测定与含量均匀度所用方法不同时的校正,以及测定结果的判断,均按现行版《中国药典》附录的规定。 取供试品10片(个),照各药品项下规定的方法,分别测定每片以标示量为100的相对含量X,求其均值X和标准差S以及标示量与均值之差的绝对值A(A=∣100-X∣);如A+1.80S≤15.0,即供试品的含量均匀度符合规定;若A+S>15.0,则不符合规定;若A+1.80S>15.0,且A+S≤15.0,则应另取20片(个)复试。根据初、复试结果,计算30片(个)的均值X、标准差S和标示量与均值之差的绝对值A;如A+1.45S≤15.0,即供试品的含量均匀度符合规定;若A+1.45S>15.0,则不符合规定。
的品种. 2. 用于单个制剂(片,个或支)主药含量少,辅料较. 多,目标难混匀(主药含量在5%以下)的品种; 3. 用于急救、毒剧药品或安全范围小的品种; 4. 主要适用于口服固体制剂的品种。 含量均匀度是考察制剂工艺水平的重要指标之一。以前是用装(重)量差异来衡量的。但随着科学技术的发展及临床要求,装(重)量差异检查不能满足对小剂量制剂质量控制的要求。重(装)量差异是检查主药和辅料的总限度,没有明确考察主药的均一性,而主药是起治疗作用的,尤其是对那些治疗剂量与中剂量比较接近的品种,控制其含量均匀度显得尤为重要。 除另有规定外,测定时取样数量和含量测定与含量均匀度所用方法不同时的校正,以及测定结果的判断,均按现行版《中国药典》附录的规定。 取供试品10片(个),照各药品项下规定的方法,分别测定每片以标示量为100的相对含量X,求其均值X和标准差S以及标示量与均值之差的绝对值A(A=∣100-X∣);如A+1.80S≤15.0,即供试品的含量均匀度符合规定;若A+S>15.0,则不符合规定;若A+1.80S>15.0,且A+S≤15.0,则应另取20片(个)复试。根据初、复试结果,计算30片(个)的均值X、标准差S和标示量与均值之差的绝对值A;如A+1.45S≤15.0,即供试品的含量均匀度符合规定;若A+1.45S>15.0,则不符合规定。")
17
(二)溶出度 溶出度系指药物从片剂或胶囊剂等固体制剂在规 定溶液、时间等条件下溶出的程度,以相当于标 示量的百分率表示。
它是评价药品制剂质量的一个内在指标,是一种 模拟口服固体制剂在胃肠通中的崩解和溶出的体 外试验法。 前面我们已经介绍了药物的一个重要的物理常数溶解度,溶解度的不同对于药物的吸收利用具有很大的影响,所以,测定制剂的溶出度是十分必要的 溶出度系指药物从片剂或胶囊剂等固体制剂在规定溶液、时间等条件下溶出的程度,以相当于标示量的百分率表示。它是评价药品制剂质量的一个内在指标,是一种模拟口服固体制剂在胃肠通中的崩解和溶出的体外试验法。

18
测定溶出度各国的方法也是不同的如

19
1995 年 USP 23 版收载了该法( 第3 法) ,应用于缓释制剂,特别是小丸( 或微丸) 制剂的释放度检查。该仪器装置采用两端装有筛网的透明中空圆筒。将样品置于圆筒中并将两端筛网紧固后,该装置在置于水浴中并内装有溶出介质的玻璃管中轻轻上下往复,解聚后的颗粒“悬浮”在往复运动的液流中。为部分模拟人体胃肠道环境,可用于存放不同 pH 介质的一系列玻璃管作为溶出杯。往复筒法的优点是所需介质少,易于改变 pH、减少死时间以及避免降解等。用于缓控释制剂溶出检查。
 ,应用于缓释制剂,特别是小丸( 或微丸) 制剂的释放度检查。该仪器装置采用两端装有筛网的透明中空圆筒。将样品置于圆筒中并将两端筛网紧固后,该装置在置于水浴中并内装有溶出介质的玻璃管中轻轻上下往复,解聚后的颗粒 悬浮 在往复运动的液流中。为部分模拟人体胃肠道环境,可用于存放不同 pH 介质的一系列玻璃管作为溶出杯。往复筒法的优点是所需介质少,易于改变 pH、减少死时间以及避免降解等。用于缓控释制剂溶出检查。")
20
该方法就是将制剂置于样品池中,溶出介质在泵压下通过流通池。该方法已经列入 USP36 版的第 4 法和《欧洲药典》( 第七版) 的官方标准,可用于篮法和桨法中有饱和度问题的药物。用于片剂和包衣片、栓剂、软胶囊、植入剂、微胶囊、粉末和颗粒等制剂溶出速率的测定。特别适用于缓释制剂的检测以及需要改变介质 pH 的溶出或释放度的分析,还可为难溶性药物提供恒定的漏槽条件。该方法具有与自动取样技术的天然适配性。
 的官方标准,可用于篮法和桨法中有饱和度问题的药物。用于片剂和包衣片、栓剂、软胶囊、植入剂、微胶囊、粉末和颗粒等制剂溶出速率的测定。特别适用于缓释制剂的检测以及需要改变介质 pH 的溶出或释放度的分析,还可为难溶性药物提供恒定的漏槽条件。该方法具有与自动取样技术的天然适配性。")
21
搅拌桨、溶出杯按桨法,但另用网碟组成其桨碟装置。置贴片的不锈钢网碟的结构

22
用于透皮药物运载系统的药物释放度的测定

23
篮法 篮法是通过固定制剂的位置过的尽可能重现的液-固接触层。不足之处如制剂中的黏性物质容易堵塞筛网;对溶出介质中溶解气体极其敏感以及当颗粒逸出网篮并浮在溶出介质中时会造成流苏的变动; 另外在试验中发现转篮法还存在着一个较为实际的问题: 例如篮内装得较满时,在仪器转动过程中仅仅可以能使释放的介质与篮外层的部分相接触,所以就常常不能较为客观地反映出溶出规律。

24
篮法是通过固定制剂的位置过的尽可能重现的液-固接触层。不足之处如制剂中的黏性物质容易堵塞筛网;对溶出介质中溶解气体极其敏感以及当颗粒逸出网篮并浮在溶出介质中时会造成流苏的变动; 另外在试验中发现转篮法还存在着一个较为实际的问题: 例如篮内装得较满时,在仪器转动过程中仅仅可以能使释放的介质与篮外层的部分相接触,所以就常常不能较为客观地反映出溶出规律。

25
桨法 此方法克服了篮法的诸多不足。但是该方法对搅拌桨和溶出杯几何尺寸的精度有相当高的要求,搅拌桨运动方向的微小改变都会引起溶出结果的变动,而这些结果的变动是可接受的。浆法具有极易导致样品上浮的缺点。

26
小杯法-缩小版桨法

27
溶出度应用的指导原则 1. 重点用于难溶性的药品品种一般指在水中微溶或不溶的;
2. 用于因制剂处方与上产工艺造成临床疗效不稳定的品种以及 治疗量与中毒量相接近的口服固体制剂(包括易溶性药品), 对后一种情况应控制两点溶出量(第一点不应溶出过多); 3. 检测方法的选择:转篮法,以100 r/min为主,桨法,以50 转/分钟为主。溶出量一般为45分钟达70%,第三法用于 规格小的品种; 4. 溶出介质应以水、0.1 mol/L盐酸、缓冲液(pH值3~8为主); 若介质中加适量有机溶剂如异丙醇、乙醇等或加分散助溶剂 如十二烷基硫酸钠(0.5%以下),应有文献数据,并尽量选用 低浓度,必要时应与生物利用度比对。
, 对后一种情况应控制两点溶出量(第一点不应溶出过多); 3. 检测方法的选择:转篮法,以100 r/min为主,桨法,以50 转/分钟为主。溶出量一般为45分钟达70%,第三法用于 规格小的品种; 4. 溶出介质应以水、0.1 mol/L盐酸、缓冲液(pH值3~8为主); 若介质中加适量有机溶剂如异丙醇、乙醇等或加分散助溶剂 如十二烷基硫酸钠(0.5%以下),应有文献数据,并尽量选用 低浓度,必要时应与生物利用度比对。")
28
溶出度测定程序 选择转速、介质、取样时间、取样点等 如是胶囊剂,空心胶囊的影响也应考察。
溶出度测定中首先应按规定对仪器进行校正,然后对研究的制剂溶出度测定进行方法学研究,如选择转速,介质。取样时间、取样点等。待以上条件确定后还应对该测定条件下的线性范围,溶液稳定性等进行考察;如是胶囊剂,空心胶囊的影响也应考察。 如是胶囊剂,空心胶囊的影响也应考察。

29
(三)崩解时限 崩解系指固体制剂在检查时限内全部崩解溶散或 成碎粒,除不溶性包衣材料或破碎的胶囊壳外, 应通过筛网。
应用范围:用于易溶性药物的压制片、薄膜衣片 、胶囊或滴丸剂及肠溶衣制剂的品种。 崩解系指固体制剂在检查时限内全部崩解溶散或成碎粒,除不溶性包衣材料或破碎的胶囊壳外,应通过筛网。崩解时限主要用于易溶性药物的压制片、薄膜衣片、胶囊或滴丸剂及肠溶衣制剂的品种。崩解时限检查中飘浮的制剂应尽量改用溶出度测定法测定。对糖衣片的崩解时限宜提高要求。崩解时限检查时的记述应包括介质,是否加档板具体崩解时间(不能笼统不超过规定时限)。
。")
30
(四)释放度 释放度系指口服药物从缓释制剂,控释制剂或肠溶制 剂在规定溶液中释放的速度和程度。
释放度检查所用介质,原则与溶出度相同,但控缓释 制剂至少应测定三个时间点的释放量。 作为缓释、控释口服固体制剂,释放度是必订的主要 项目 释放度系指口服药物从缓释制剂,控释制剂或肠溶制剂在规定溶液中释放的速度和程度。用于控释与缓释制剂,按《中国药典》附录释放度第一法检查。用于肠溶制剂,按《中国药典》附录释放度第二法检查。 第一法检查 照溶出度测定法项下进行,但一般采用三个时间取样,在规定时间、规定取样点,吸取溶液适量,立即经0.8μm微孔滤膜滤过,自取样至滤过应在30秒钟内完成,并及时补充所耗的溶剂。取滤液,照各药品项下规定的方法测定,算出每片(个)的释放量。 第二法 用于肠溶制剂 酸中释放量 除另有规定外,量取0.1mol/L盐酸溶液750ml,注入每个容器,加温使溶液温度保持在37±0.5℃,调整转速并保持稳定,取6片(个)分别投入转篮或容器中,按各药品项下规定的方法,开动仪器运转2小时,立即在规定取样点吸取溶液适量,立即经0.8μm微孔滤膜滤过,自取样至滤过应在30秒钟内完成,滤液按各药品项下规定的方法测定,算出每片(个)的酸中释放量。 缓冲液中释放量 上述酸液中加入0.2mol/L磷酸钠溶液250ml(必要时用2mol/L盐酸溶液或2mol/L氢氧化钠溶液调节pH至6.8±0.05)继续运转45分钟,或按各药品项下规定的时间,在规定取样点吸取溶液适量,立即经0.8μm微孔滤膜滤过,自取样至滤过应在30秒钟内完成,滤液按各药品项下规定的方法测定,算出每片(个)的缓冲液中释放量。 而第三法是用于透皮帖剂的,用的是桨蝶法进行的测定。 除另有规定外,取样数量和对测定结果的判断均按现行版《中国药典》附录的规定。实验操作中注意事项与有关要求均同溶出度项下,研究新药的释放度应在实验条件下,有每片(粒)供试品在各个时间点的测定数值,作为缓释、控释口服固体制剂,释放度是必订的主要项目。
的释放量。 第二法. 用于肠溶制剂. 酸中释放量. 除另有规定外,量取0.1mol/L盐酸溶液750ml,注入每个容器,加温使溶液温度保持在37±0.5℃,调整转速并保持稳定,取6片(个)分别投入转篮或容器中,按各药品项下规定的方法,开动仪器运转2小时,立即在规定取样点吸取溶液适量,立即经0.8μm微孔滤膜滤过,自取样至滤过应在30秒钟内完成,滤液按各药品项下规定的方法测定,算出每片(个)的酸中释放量。 缓冲液中释放量. 上述酸液中加入0.2mol/L磷酸钠溶液250ml(必要时用2mol/L盐酸溶液或2mol/L氢氧化钠溶液调节pH至6.8±0.05)继续运转45分钟,或按各药品项下规定的时间,在规定取样点吸取溶液适量,立即经0.8μm微孔滤膜滤过,自取样至滤过应在30秒钟内完成,滤液按各药品项下规定的方法测定,算出每片(个)的缓冲液中释放量。 而第三法是用于透皮帖剂的,用的是桨蝶法进行的测定。 除另有规定外,取样数量和对测定结果的判断均按现行版《中国药典》附录的规定。实验操作中注意事项与有关要求均同溶出度项下,研究新药的释放度应在实验条件下,有每片(粒)供试品在各个时间点的测定数值,作为缓释、控释口服固体制剂,释放度是必订的主要项目。")
31
(五)有关物质 原料药 稳定 制剂加工工艺 稳定性试验 有关物质增加 制剂质量标准√ 原料与制剂都比较稳定 原料药项下√制剂质量标准×
有关物质研究是药品质量研究中关键性的项目之一,其含量是反映药品纯度的直接指标。对药品的纯度要求,应基于安全性和生产实际情况两方面的考虑, 制剂在工艺过程与贮藏过程均应对有关物质进行考察。在有关物质的含义与检测方法均与原料药项下相同外,还应考察制剂过程有关物质的增加情况。在经过制剂加工工艺后如果有关物质增加或经稳定性试验结果制剂比原料药有关物质增加,则应在制剂质量标准中作出规定。如果经过以上各项考察原料与制剂都比较稳定,则有关物质检查在原料药项下作控制规定。制剂不一定再订此项,但有的原料药本身不够稳定,制剂过程虽无明显增加有关物质,但放置过程如同原料一样会有变化,则在制剂中检查有关物质就非常必要,如盐酸雷尼替丁及其制剂中有关物质的检查,制剂中有关物质检查方法基本同原料药。但要研究制剂辅料对有关物质检查的干扰,并应设法排除干扰。 原料药 不稳定 制剂加工工艺 无增加有关物质 经放置,如同原料一样会有变化 制剂质量标准√

32
(六)pH值 pH值是注射剂必须控制的项目。有的品种对不同 pH值影响较大的,其范围应从严控制。
水6,

33
(七)注射液中不溶性微粒检查 装量在100 ml以上的供静脉滴注用注射液,在澄 明度检查符合规定后,再检查不溶性微粒方法与 限度规定均按《中国药典》附录。
注射液中不溶性微粒检查 装量在100 ml以上的供静脉滴注用注射液,在澄 明度检查符合规定后,再检查不溶性微粒方法与 限度规定均按《中国药典》附录。")
34
(八)异常毒性检查 有的注射液必要时要检查异常毒性,过敏试验、降压 物质、热原,细菌内毒素等项:方法均按《中国药典 》附录,但热原剂量(即家兔体重每1 kg注射多少)要经 实验探索,或参考国外药典及有关文献。 前面我已经对其进行了介绍,现在就不再仔细介绍了

35
(九)微生物限度检查 在研究新药时对口服固体制剂均应作此项检查, 应符合《中国药典》规定。
微生物限度检查法系检查非规定灭菌制剂及其原料、辅料受微生物污染程度的方法。检查项目包括细菌数、霉菌数、酵母菌数及控制菌检查。在研究新药时对口服固体制剂均应作此项检查,应符合《中国药典》规定。 除另有规定外,一般供试品的检验量为10g 或10ml;膜剂为100c㎡;贵重药品、微量包装药品的检验量可以酌减。要求检查沙门菌的供试品,其检验量应增加20g 或20ml(其中10g或者10ml 用于阳性对照试验)。 液体供试品 取供试品10ml,加pH7.0 无菌氯化钠-蛋白胨缓冲液至100ml,混匀,作为1∶10 的供试液。油剂可加入适量的无菌聚山梨酯80 使供试品分散均匀。水溶性液体制剂也可用混合的供试品原液作为供试液。 固体、半固体或粘稠性供试品 取供试品10g,加pH7.0 无菌氯化钠-蛋白胨缓冲液至100ml,用匀浆仪或其他适宜的方法,混匀,作为1∶10 的供试液。必要时加适量的无菌聚山梨酯80,并置水浴中适当加温使供试品分散均匀。 3.需用特殊供试液制备方法的供试品 (1)非水溶性供试品 (2)膜剂供试品 (3)肠溶及结肠溶制剂供试品 取供试品10g,加pH6.8 无菌磷酸盐缓冲液(用于肠溶制剂)或pH7.6 无菌磷酸盐缓冲液(用于结肠溶制剂)至100ml,置45℃水浴中,振摇,使溶解,作为1∶10 的供试液。 (4)气雾剂、喷雾剂供试品 (5)贴膏剂供试品 (6)具抑菌活性的供试品 当供试品有抑菌活性时,采用下列方法进行处理,以消除供试液的抑菌活性后,再依法检查。 ①培养基稀释法 取规定量的供试液,至较大量的培养基中,使单位体积内的供试品含量减少,至不含抑菌作用。测定细菌、霉菌及酵母菌的菌数时,取同稀释级的供试液2ml,每1ml 供试液可等量分注多个平皿,倾注琼脂培养基,混匀,凝固,培养,计数。每1ml 供试液所注的平皿中生长的菌数之和即为1ml的菌落数,计算每1ml 供试液的平均菌落数,按平皿法计数规则报告菌数;控制菌检查时,可加大增菌培养基的用量。 ② 离心沉淀法 取一定量的供试液, 500 转/分离心3 分钟,取全部上清液混合,用于细菌检查。 ③薄膜过滤法 见细菌、霉菌及酵母菌计数项下的“薄膜过滤法”。 ④中和法 凡含汞、砷或防腐剂等具有抑菌作用的供试品,可用适宜的中和剂或灭活剂消除其抑菌成分。中和剂或灭活剂可加在所用的稀释液或培养基中。
。 液体供试品. 取供试品10ml,加pH7.0 无菌氯化钠-蛋白胨缓冲液至100ml,混匀,作为1∶10 的供试液。油剂可加入适量的无菌聚山梨酯80 使供试品分散均匀。水溶性液体制剂也可用混合的供试品原液作为供试液。 固体、半固体或粘稠性供试品. 取供试品10g,加pH7.0 无菌氯化钠-蛋白胨缓冲液至100ml,用匀浆仪或其他适宜的方法,混匀,作为1∶10 的供试液。必要时加适量的无菌聚山梨酯80,并置水浴中适当加温使供试品分散均匀。 3.需用特殊供试液制备方法的供试品. (1)非水溶性供试品. (2)膜剂供试品. (3)肠溶及结肠溶制剂供试品. 取供试品10g,加pH6.8 无菌磷酸盐缓冲液(用于肠溶制剂)或pH7.6 无菌磷酸盐缓冲液(用于结肠溶制剂)至100ml,置45℃水浴中,振摇,使溶解,作为1∶10 的供试液。 (4)气雾剂、喷雾剂供试品. (5)贴膏剂供试品. (6)具抑菌活性的供试品. 当供试品有抑菌活性时,采用下列方法进行处理,以消除供试液的抑菌活性后,再依法检查。 ①培养基稀释法 取规定量的供试液,至较大量的培养基中,使单位体积内的供试品含量减少,至不含抑菌作用。测定细菌、霉菌及酵母菌的菌数时,取同稀释级的供试液2ml,每1ml 供试液可等量分注多个平皿,倾注琼脂培养基,混匀,凝固,培养,计数。每1ml 供试液所注的平皿中生长的菌数之和即为1ml的菌落数,计算每1ml 供试液的平均菌落数,按平皿法计数规则报告菌数;控制菌检查时,可加大增菌培养基的用量。 ② 离心沉淀法 取一定量的供试液, 500 转/分离心3 分钟,取全部上清液混合,用于细菌检查。 ③薄膜过滤法 见细菌、霉菌及酵母菌计数项下的 薄膜过滤法 。 ④中和法 凡含汞、砷或防腐剂等具有抑菌作用的供试品,可用适宜的中和剂或灭活剂消除其抑菌成分。中和剂或灭活剂可加在所用的稀释液或培养基中。")
36
请在此处输入您的标题 非水溶性供试品 膜剂供试品 肠溶及结肠溶 制剂供试品 气雾剂、喷雾 剂供试品 具抑菌活性的 供试品 贴膏剂供试品

37
(十)有机溶剂残留量检查 制剂工艺用了有毒的有机溶剂,应检查有机溶剂 残留量:方法研究同原料药项下。对于一类、二 类供静脉注射的新药,“处方”中如加有抗氧剂、 络合剂、防腐剂等均应作定量测定。 因有机溶剂易挥发性,一般用气相色谱法进行检测。 顶空毛细管气相色谱法测定茶多酚中正己烷、氯仿、乙酸乙酯和丙酮四种有机溶剂残留量

38
原料药及制剂检查 原料药检测项目 杂质检查及限量的基本原则 制剂检查的内容 溶出度 崩解时限 释放度

Similar presentations

 导致胎儿缺氧的母体因素有 ①微小动脉供血不足:如妊高征等 ②红细胞携氧量不足:如重度贫血、一氧化碳中毒等;>")
列举植物的感应性现象 a 2 )描述植物生长素发现的历史, 体验科学发现的过程 a 2 、人体生命活动的调节 列举激素对人体生命活动调节 的作用 a.>")








 血液中转氨酶活力的测定 一. 目的 : 了解转氨酶在代谢过程中的重要作用及其在临 床诊断中的意义, 学习转氨酶活力测定的原理和方 法。 二. 原理 : 生物体内广泛存在的氨基转换酶也称转氨酶, 能 催化 α – 氨基酸的 α – 氨基与 α – 酮基互换, 在氨基酸 的合成和分解尿素和嘌呤的合成等中间代谢过程中.>")








