Download presentation
Presentation is loading. Please wait.

1
國內實施BE臨床試驗情形及其可靠性驗證與藥品品質之探討
鮑力恒 博士 台灣藥物品質協會 秘書長 國防醫學院藥學系副教授

2
本尊與分身? 原廠藥 學名藥

3
學名藥查驗登記

4
我國學名藥的審查 藥品查驗登記審查準則:與國內已核准之藥品具 安全性、有效性 -生體相等性試驗 (Bioequivalence, BE)
- 同成分、 - 同劑型、 - 同劑量、 - 同療效之製劑 安全性、有效性 -生體相等性試驗 (Bioequivalence, BE) 品質 -原料及成品檢驗規格方法成績書 -製程管制 -安定性資料 -送驗
 品質. -原料及成品檢驗規格方法成績書. -製程管制. -安定性資料. -送驗.")
5
我國學名藥之管理 上市前-生體相等性審查 -依據「藥品生體可用率及生體相等性試驗準則」 上市後 配方與製程修改
依據「藥品查驗登記審查準則 」第46條、第56條,應檢附生體相等性或溶離率試驗報告(<10%以內修改) cGMP查廠及PIC/S GMP之推動 療效不相等通報系統(建制中-藥害救濟基金會)
 cGMP查廠及PIC/S GMP之推動. 療效不相等通報系統(建制中-藥害救濟基金會)")
6
品質之管制

7
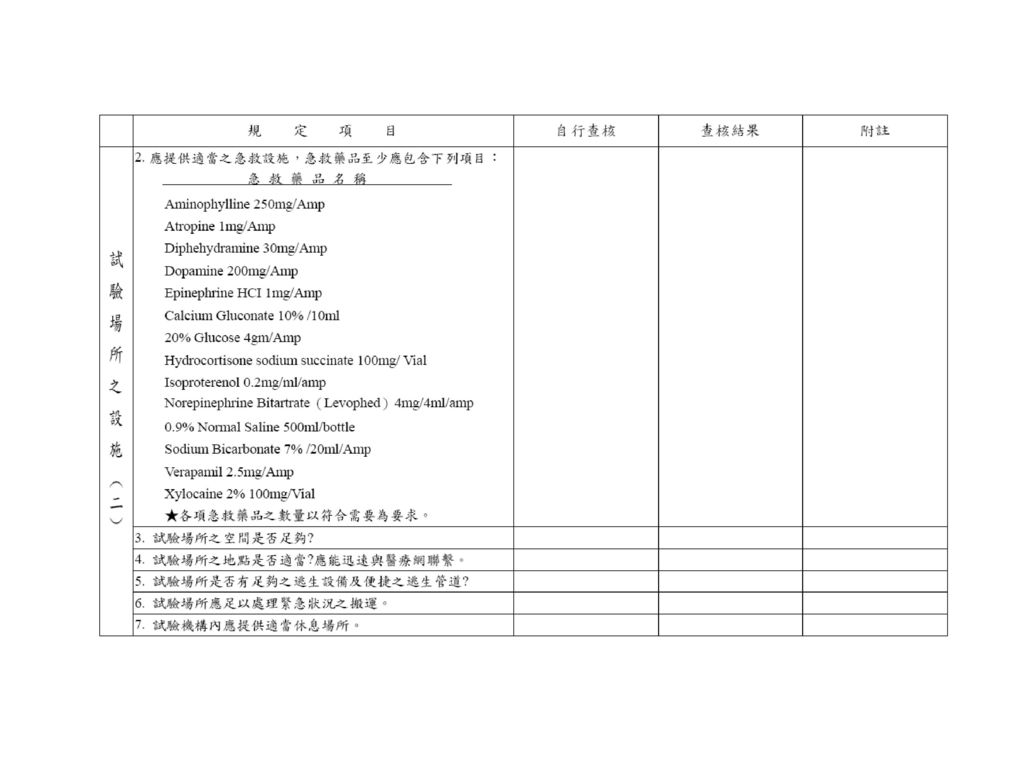
體外品管之指標 藥品含量 藥品主成份含量均一度 藥品之安定性 藥品之溶離試驗等 符合藥典之規定

8
? A藥 = B藥 A藥 = B藥 療效及安全性

10
體內品管之指標 – 生體可用率 藥品有效成份由製劑中吸收進入全身血液循環或作用部位之量與速率之指標。

11
體內品管之指標 – 生體可用率 (BA) 最高濃度:吸收之速率 血漿中藥物濃度 曲線下之面積: 吸收進入體內之量 時間
 最高濃度:吸收之速率 血漿中藥物濃度 曲線下之面積: 吸收進入體內之量 時間")
12
生體相等性 (BE) 生體相等性(Bioequivalence): 指二個藥劑相等品或藥劑替代品 ,於適當研究設計下,以相同條件、相同莫耳劑量(molar dose) 給與人體時,具有相同之生體可用率。
 生體相等性(Bioequivalence): 指二個藥劑相等品或藥劑替代品 ,於適當研究設計下,以相同條件、相同莫耳劑量(molar dose) 給與人體時,具有相同之生體可用率。")
13
生體相等性 (BE) 以相同條件給予同一組人: 隨機交叉試驗方式,以減少 個體間之差異 試驗期一 試驗期二 第一組試驗者 A 藥 B 藥
第二組試驗者

14
生體相等性 (BE) - 原製造廠 - 非原製造廠 血漿中藥物濃度 時間
 - 原製造廠 - 非原製造廠 血漿中藥物濃度 時間")
15
生體相等性 (BE) 之判斷 時間 時間 時間 血漿中藥物濃度 血漿中藥物濃度 血漿中藥物濃度 無統計上顯著差異
90% 信賴區間: % Cmax 及 AUC 血漿中藥物濃度 時間

16
BE 試驗的設計與執行 藥品生體可用率及生體相等性試驗準則

17
藥品生體可用率及生體相等性試驗準則 中華民國98年4月2日制(訂)定
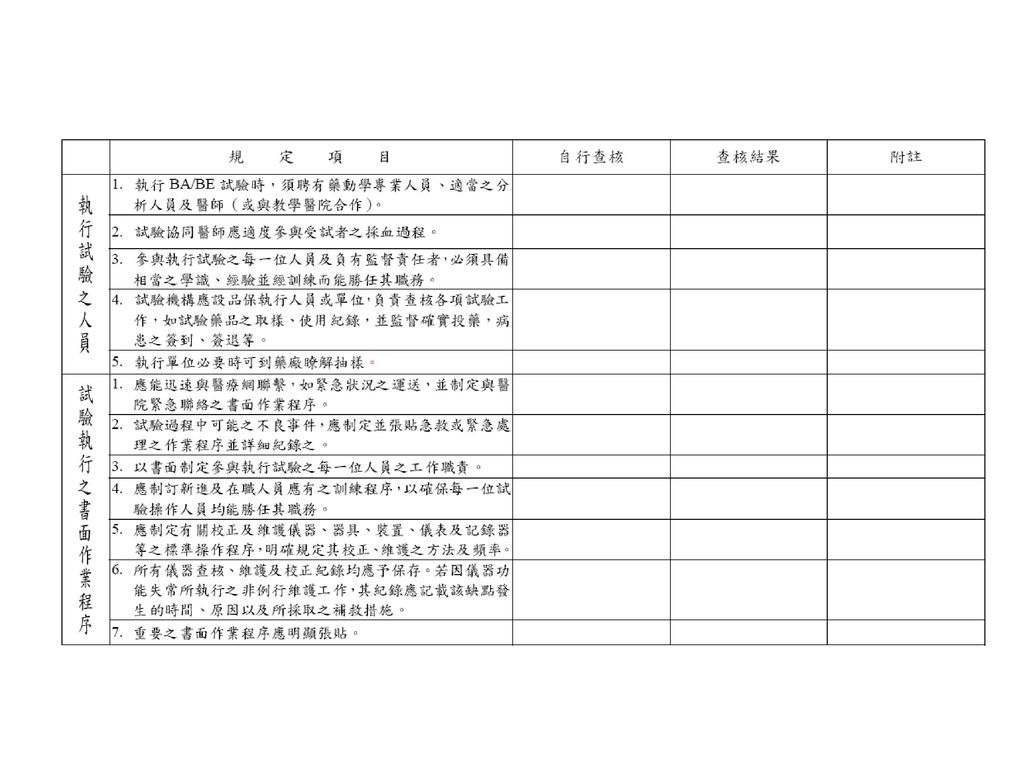
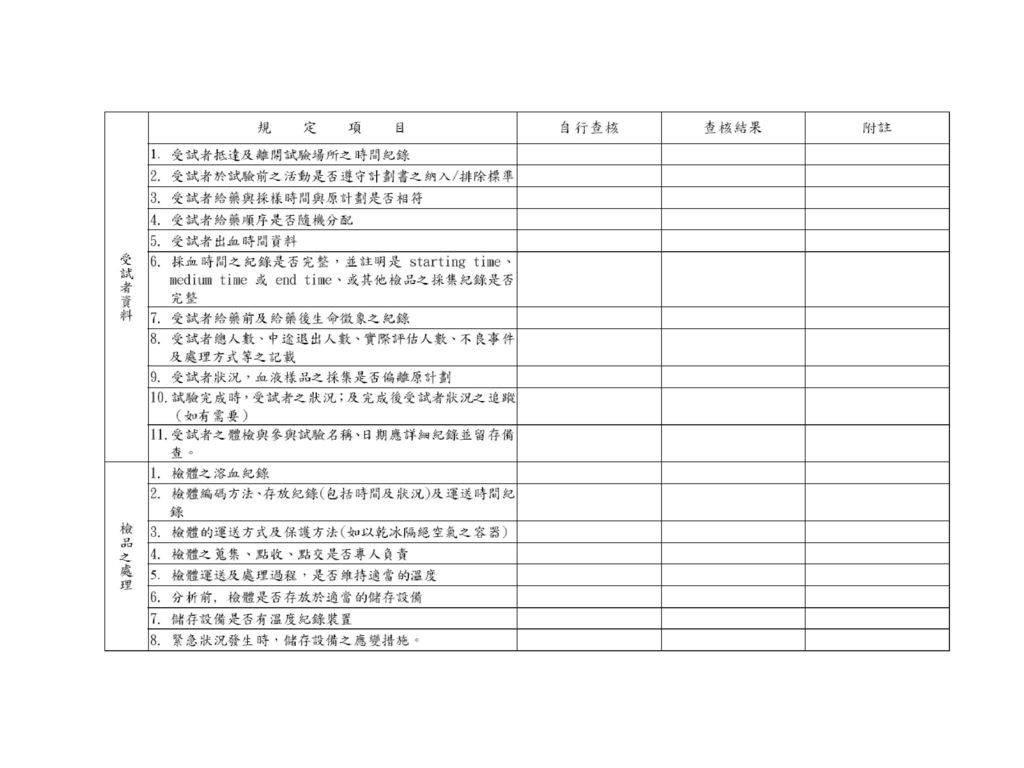
受試者之人數,至少應有十二名,且應用適當之檢定力計算( appropriate power calculations)以評估受試者之人數。 試驗進行前,應經 人體試驗委員會之同意,並取得受試者同意書。
以評估受試者之人數。 試驗進行前,應經 人體試驗委員會之同意,並取得受試者同意書。")
18
藥品生體可用率及生體相等性試驗準則 對照藥選擇 絕對生體可用率:IV 相對生體可用率:不同劑型、劑量、投與途徑 生體相等性試驗 藥劑相等品-同成分、同劑型、同劑量、不同成分 劑量選擇 生體可用率試驗:單一劑量單次使用、多次使用 逐步上升劑量 生體相等性試驗:IR-單劑量 SR-多劑量;單劑量+food effect 受試者選擇 健康志願者/病患 性別

19
試 驗 設 計 隨機雙向、多向、拉丁方塊交叉試驗 Phase I II Group1 A→B Group2 B→A 隨機平行試驗
受試者人數 至少在12名(含)以上 禁服藥物、食物 試驗前禁服藥物兩週以上,服藥前禁食十小時以上,服藥 後禁食四小時。
以上. 禁服藥物、食物. 試驗前禁服藥物兩週以上,服藥前禁食十小時以上,服藥. 後禁食四小時。")
20
試 驗 設 計 評估標的 a.血液、尿液中藥物或其代謝物濃度 b.適當藥理反應或臨床效應 採樣時間 a.血液:最高血中濃度後三倍以上排除半衰期 b.尿液:排除半衰期七倍以上 c.採樣次數:足可說明藥品於體內之吸收、分佈、排除相 洗除時間(washout period) a.第一次用藥最後採樣到第二次用藥的間隔時間 b.藥品排除半衰期之五倍 c. 服藥期血中濃度不超過Cmax之5%
 a.第一次用藥最後採樣到第二次用藥的間隔時間 b.藥品排除半衰期之五倍 c. 服藥期血中濃度不超過Cmax之5%")
21
藥物分析 (依國際規範) 分析方法 a.測得原始試驗藥品或其代謝物 b.有適當之最低可測得濃度 c.以代謝物為分析標的者,應事先經中央衛生主管機關認可 分析方法確效(Bioanalytical Method Validation) a.精密度、準確度、選擇性、基質效應、最低定量濃度、系統適用性 b.檢體之安定性 冷凍與解凍、短期室溫、長期 安定性、儲液安定性、分析期間安定性
 a.精密度、準確度、選擇性、基質效應、最低定量濃度、系統適用性 b.檢體之安定性 冷凍與解凍、短期室溫、長期 安定性、儲液安定性、分析期間安定性")
22
評 估 參 數 血中藥物濃度 速放劑型 單劑量試驗:Cmax、AUC0-t、AUC0-8、Tmax、T½、MRT 多劑量試驗:Cmax,ss 、AUC0- t 、Fluctuation、 Tmax 、Cmin 控釋劑型 單劑量試驗+食物影響試驗: Cmax 、 AUC0-t 、 AUC0-8…. 多劑量試驗: Cmax,ss 、 AUC0-t 、 Fluctuation 、 Tmax 、 Cmin 尿液藥物濃度 藥在各時段的排出量 起始至終止時間的累積量

23
計畫書撰寫 Ⅴ藥品分析 a.分析檢測物、標的物 b.分析方法 c.最低準確可測定濃度 Ⅵ數據分析 a.藥動學參數 b.統計分析方法
Ⅰ藥物背景資料 Ⅱ試驗目的 Ⅲ研究人員&機構 Ⅳ試驗計劃 a.受試者選擇 b.試驗執行步驟 c.試驗藥物 d.投藥計劃 e.簡品採集量/時間 f.簡品運送及儲存 g.參與試驗風險 h.處置與追蹤 i.同意書籤署 j.藥品管理

24
BE執行時之事前控管 CRO或Sponsor必須事先通知TFDA未來兩個月預計進行之案件 受試者資料須在網路平台登錄及比對確認
目前此由中華民國製藥發展協會負責管理

25
藥品臨床試驗受試者資格確認平台 中華民國製藥發展協會 98/10/01 正式上線

26
執行試驗品質與受試者保護 受試者重覆受試問題 彙整受試者資訊 DOH CROs CROs CROs 受試者健康 Sponsors CROs
??? Sponsors CROs CROs CROs

27
藥品臨床試驗受試者資格確認平台依據 為依據「人體試驗倫理相關規範」及行政院衛生署公告「受試者之臨床試驗間隔,應符合捐血者健康標準之規定:
每次捐血250毫升者,其捐血間隔應為2個月以上; 每次捐血500毫升者,其捐血間隔應為3個月以上」, 以保障受試者之健康安全,特建立「藥品臨床試驗受試者資格確認 平台」。

28
受試者資格確認平台-主畫面

29
受試者試驗日期登錄

30
受試者試驗日期登錄報告

31
受試者試驗紀錄查詢結果_1

32
受試者試驗紀錄查詢結果_2

33
受試者資料確認列印

34
BE報告的審查 每一報告都須經衛生署審查 衛生署收件 依照申請表格(TFDA)初審 送交BE委員複審 製造批次紀錄 試驗藥品的照片及溶離試驗
IRB同意函 臨床試驗執行日期查驗(需事先通報TFDA) 受試者體檢資料,臨床試驗紀錄 分析確效(validation)的所有圖譜 至少1/3臨床檢體分析的圖譜 統計分析資料(TFDA在做確認) 試驗藥品留樣五年(報告經衛生署同意後)
 受試者體檢資料,臨床試驗紀錄. 分析確效(validation)的所有圖譜. 至少1/3臨床檢體分析的圖譜. 統計分析資料(TFDA在做確認) 試驗藥品留樣五年(報告經衛生署同意後)")
35
生體相等性 (BE) 之判斷 實驗設計 受試者人數 血中藥物濃度分析方法 依統計學原理來計算 最高濃度 曲線下之面積 是否符合基準規定
 之判斷 實驗設計 受試者人數 血中藥物濃度分析方法 依統計學原理來計算 最高濃度 曲線下之面積 是否符合基準規定")
36
後續追蹤查核 衛生署不定期查核 95年起執行無預警查核 執行臨床試驗場所 分析場所(CRO公司) 查核原始資料與BE報告的一致度及
釐清問題或疑點

37
藥品BA/BE試驗無預警查核作業流程 (參照藥品BA/BE試驗場所或實驗室查核會議進行之流程 CRO公司通知未來兩個月進行案件 衛生署
查核小組 確定BA/BE試驗查核日期、時間、地點 安排查核小組成員 行文通知並確認 會合地點、時間 (參照藥品BA/BE試驗場所或實驗室查核會議進行之流程 查核作業之進行 完成查核觀察報告

38
後續監控及追蹤 藥品療效不等通報作業 財團法人藥害救濟基金會建置中
後續監控及追蹤 藥品療效不等通報作業 財團法人藥害救濟基金會建置中

39
藥品療效不等通報作業 建置通報系統收集藥品療效不相等 通報案件 系統性尋找有潛在問題之產品 維護學名藥之療效與安全性

40
通報作業流程

41
通報方式 通報機構:財團法人藥害救濟基金會 地址:台北市羅斯福路一段32號2F 傳真:23584098;23584100

42
提升原料藥品質管理 Drug Master File (DMF)原料藥主檔案 TFDA核准 台灣藥物品質協會(TPQRI)協助建置中
98年09月30日衛署藥字第 號公告 TFDA核准 電腦建檔其DMF詳細資料 包括國產、輸入及自用原料藥。 製劑廠變更原料藥供應商得向衛生署申請變更登記 台灣藥物品質協會(TPQRI)協助建置中 (Taiwan Product Quality Research Institute)
協助建置中. (Taiwan Product Quality Research Institute)")
43
BE臨床試驗可靠性驗證與藥品品質管控 cGMP PIC/S GMP QC 藥典等規範 藥品BA/BE試驗準則 專家審查 無預警查核
藥品療效不等通報 藥品製造 試驗執行 BE審核 上市後監控 原料藥主檔案 事前 報備 受試者登錄 TFDA 查驗比對

44
品質之路

Similar presentations
















。>")



