Download presentation
1
溶血性贫血

2
溶血性贫血的定义 溶血性贫血(hemolytic anemia)是指由于红细胞寿命缩短、破坏增加、骨髓造血功能不足以代偿红细胞的耗损
溶血性疾病:发生溶血,骨髓能够代偿(6~8倍能力),不出现贫血
,不出现贫血.")
3
溶贫的分类 1.按发病和病情分:急性、慢性 2.按发生场所分:血管内、血管外 3.按病因和发病机制分类(最有价值) (1)红细胞自身缺陷
(2)外部因素所致溶血
外部因素所致溶血.")
4
1.红细胞膜缺陷——多为遗传病 红细胞内在因素损伤 血影蛋白:由结构相似的α链、β链组成异二聚体,两个二聚体头与头相接连形成四聚体。 锚蛋白
带3蛋白 血型糖蛋白 带4.1蛋白 带4.2蛋白 肌动蛋白

5
垂直作用异常会减少红细胞表面积,导致球形红细胞出现。
垂直的相互作用(Vertical interaction) 垂直于红细胞膜平面,参与骨架网络结构域整体膜成分的附连 a. 血影蛋白—锚蛋白—带3蛋白接触 b. 带4.2蛋白辅助带3蛋白与锚定蛋白结合
 垂直于红细胞膜平面,参与骨架网络结构域整体膜成分的附连. a. 血影蛋白—锚蛋白—带3蛋白接触. b. 带4.2蛋白辅助带3蛋白与锚定蛋白结合.")
6
平行作用异常会导致椭圆形红细胞出现。 平行的相互作用(Horizontal interaction)
平行于红细胞膜平面,维持膜下骨架网络结构的双向连续性,使红细胞膜获得高强度的抗张力。 a. 血影蛋白异质二聚体的相互接触(SpD-SpD) b. SpD-蛋白—带4.1蛋白—肌动蛋白
 b. SpD-蛋白—带4.1蛋白—肌动蛋白.")
7
HS 遗传性球形红细胞增多症 HE 遗传性椭圆形红细胞增多症 受累膜蛋白 疾病 解释 锚蛋白 HS 典型HS最常见突变 带3蛋白 血影蛋白α
7 受累膜蛋白 疾病 解释 锚蛋白 HS 典型HS最常见突变 带3蛋白 血影蛋白α HS,HE 典型HE最常见突变 血影蛋白β 带4.2蛋白 带4.1蛋白 HE HS 遗传性球形红细胞增多症 HE 遗传性椭圆形红细胞增多症

8
8

9
磷酸己糖旁路+GSH代谢 2.红细胞 酶缺陷 糖酵解途径

10
红细胞酶缺陷

11
红细胞酶缺陷

12
3.珠蛋白异常 1)珠蛋白合成异常——地中海贫血 2)异常血红蛋白病 镰状细胞贫血 不稳定血红蛋白病 血红蛋白M病 氧亲和力异常血红 蛋白病
珠蛋白合成异常——地中海贫血 2)异常血红蛋白病 镰状细胞贫血 不稳定血红蛋白病 血红蛋白M病 氧亲和力异常血红 蛋白病")
13
珠蛋白生成障碍性贫血

14
β地中海贫血 1.静止型携带者 2.轻型贫血症状 3.中间型贫血症状 4.重型β地中海贫血 5. β珠蛋白生成障碍型贫血复合β珠蛋白链结构异常

15
红细胞外在因素损伤

16
自身免疫性溶血性贫血 免疫性溶血 免疫性溶血是指由于免疫反应而使红细 胞破坏加速,寿命缩短的过程。
免疫性溶血 免疫性溶血是指由于免疫反应而使红细 胞破坏加速,寿命缩短的过程。 溶血性贫血 溶血性贫血是由于红细胞破坏增多、增 速, 超过造血补偿能力范围,所发生的一种贫血。 自身免疫性溶血性贫血 是由于机体免疫功能异常,产生了抗自身红细胞的抗体,使红细胞破坏加速造成的溶血性贫血,在后天性溶血性贫血中多见。

17
自身免疫性溶血性贫血按照抗体分型: 1、 温抗体型自身免疫溶血性贫血(WAIHA) 原发性的 (自发性) 继发性的 (与淋巴瘤、系统性红斑狼疮、 癌症或药物处理有关) 2、 冷凝集素综合征(CAS) 原发性的 (自发的) 继发性的 (与淋巴瘤支原体肺炎、传染性 单核细胞增多症 有关)
 原发性的 (自发的) 继发性的 (与淋巴瘤支原体肺炎、传染性 单核细胞增多症 有关)")
18
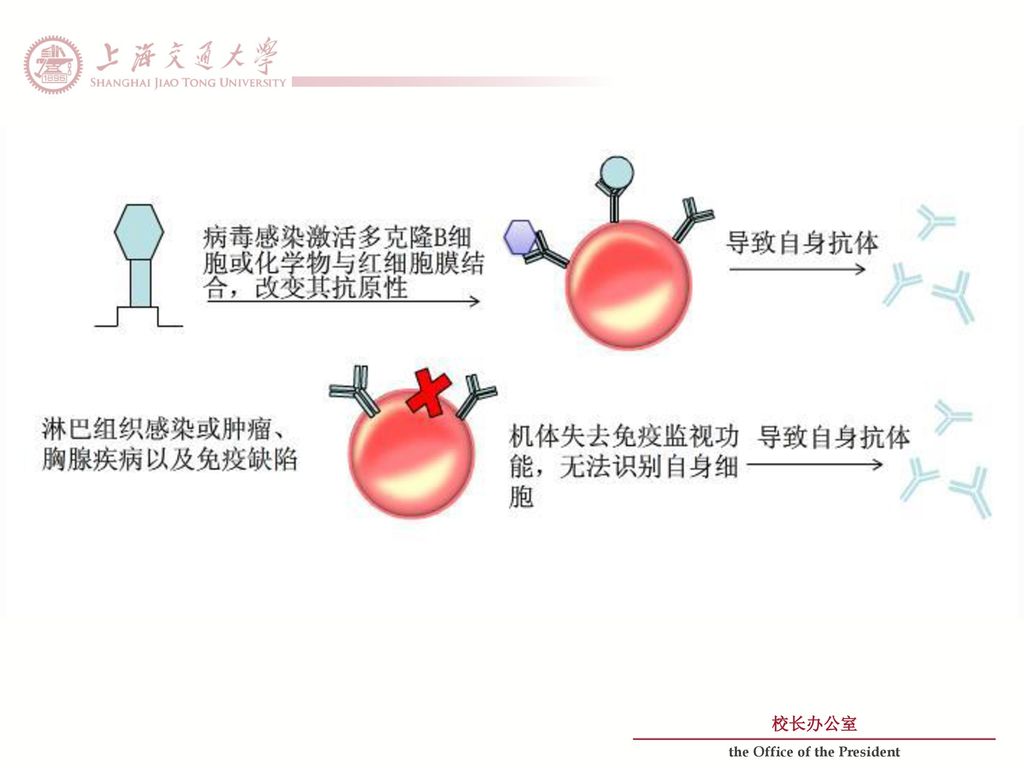
病毒感染激活多克隆B细胞或化学物与红细胞膜结合,改变其抗原性 淋巴组织感染或肿瘤、胸腺疾病以及免疫缺陷
发病机制 病毒感染激活多克隆B细胞或化学物与红细胞膜结合,改变其抗原性 导致自身抗体 淋巴组织感染或肿瘤、胸腺疾病以及免疫缺陷 机体失去免疫监视功能,无法识别自身细胞 导致自身抗体 导致自身抗体

19
发病机制 自身免疫耐受异常—DC调控异常、T和B细胞免疫耐受异 常
免疫调节异常—包括Th1/Th2细胞组成的细胞和体液免疫 调节“枢纽”以及“调节性T细胞” 抗体后调节异常 基因易感性—早年动物研究显示,存在2个优势即Aia1和Aia 2;新近发现7号染色体上的Aia3和1号染色体上的Nba2基 因与发病有关。人类Fas基因缺失,可使自身抗体明显增加 而导致AIHA。CTL相关抗原4(CTLA-4)的第49外显子A→T 突变与AIHA有明显相关性
的第49外显子A→T 突变与AIHA有明显相关性.")
20
新生儿溶血病 【发病机制】 胎儿血因某种原因进入母体,由父亲方面遗传来的显性抗原导致母体产生相应的IgM抗体。当胎儿血再次进入母体,母体发生次发免疫反应。 免疫反应中产生大量IgG抗体,此抗体又可通过胎盘进入胎儿血循环中导致胎儿、新生儿体内特异性抗原抗体反应,使红细胞致敏单核-吞噬细胞系统内破坏而溶血。

21
新生儿溶血病 1、ABO血型不合溶血病 A或B型母亲的天然抗A或抗B抗体主要为不能通过胎盘的IgM抗体,而存在于O型母亲中的同种抗体以IgG为主,因此ABO溶血病主要见于O型母亲、A或B型胎儿。抗A或抗B抗体大部分被其他组织和血浆中的可溶性A和B血型物质的中和和吸收,发病者仅占少数。

22
新生儿溶血病 2、Rh血型不合溶血病 初次免疫反应产生IgM抗体需要2~6个月,且较弱不能通过胎盘进入胎儿体内,而胎儿红细胞进入母体多数发生在妊娠末期或临产时,故第一胎常处于初次免疫反应的潜伏阶段。 再次妊娠第2次发生免疫反应时,仅需数天就可出现,主要为IgG能通过胎盘的抗体,并能迅速增多,故往往第二胎才发病。Rh系统的抗体只能由人类红细胞引起,若母亲有过输血史,且Rh血型又不合,或外祖母为Rh阳性,母亲出生前已被致敏,则第一胎也可发病。

23
输血性溶血 供血者血清中抗体——结合——受血者血细胞 受血者血清中抗体——结合——供血者血细胞 溶血的发生

24
药物性溶血 本病是因为药物进入机体后,由于免疫等因素从而引起红细胞大量破坏,临床上出现贫血、黄疸、酱油色尿等溶血表现。药物对造血系统的影响,溶血性贫血相对较少见,仅占10%,但急性溶血有生命危险。根据发病机理不同,药物性溶血性贫血可分为以下三种 (1)药物性免疫,导致有抗体介导的溶血反应 (2)药物作用于遗传性酶缺陷的红细胞(如G6PD缺乏者) (3)药物对异常血红蛋白所致的溶血反应。
药物性免疫,导致有抗体介导的溶血反应. (2)药物作用于遗传性酶缺陷的红细胞(如G6PD缺乏者) (3)药物对异常血红蛋白所致的溶血反应。")
25
引起红细胞G6PD缺乏者溶血的药物 抗疟药磺胺类解热镇痛药类其他第Ⅰ类:肯定能引起溶血的药物(共22种) 伯氨喹啉、磺胺甲嘧啶等
 伯氨喹啉、磺胺甲嘧啶等")
26
物理性溶血 人造瓣膜,微血管凝血性病变 血细胞通过时受到物理损伤,结构破坏

27
物理性溶血 大面积烧伤(高温破坏红细胞结构) 行军性血红蛋白尿
 行军性血红蛋白尿")
28
毒素和生物性溶血 化学因素:砷化物,铅,药物等 生物因素:蛇毒,疟疾,黑热病等

29
脾功能亢进 单核细胞系统功能亢进——导致红细胞的破坏。

30
临床表现 患者的临床表现主要取决于溶血的场所、程度、速率等及持续的时间以及心肺的代偿能力和基础病。 急性溶血多为血管内溶血,发病急骤。
(1)寒战、发热、头痛、呕吐、四肢腰背疼痛及腹痛、血红蛋白尿、甚至休克。 (2)黄疸 (3)贫血
寒战、发热、头痛、呕吐、四肢腰背疼痛及腹痛、血红蛋白尿、甚至休克。 (2)黄疸. (3)贫血.")
31
慢性溶血多为血管外溶血,发病缓慢,症状较轻
特征:贫血、黄疸、脾大。 一般患者对贫血有较好代偿,症状较轻 可并发胆石症、肝功能损害。 再生障碍性危象。

33
实验室诊断

34
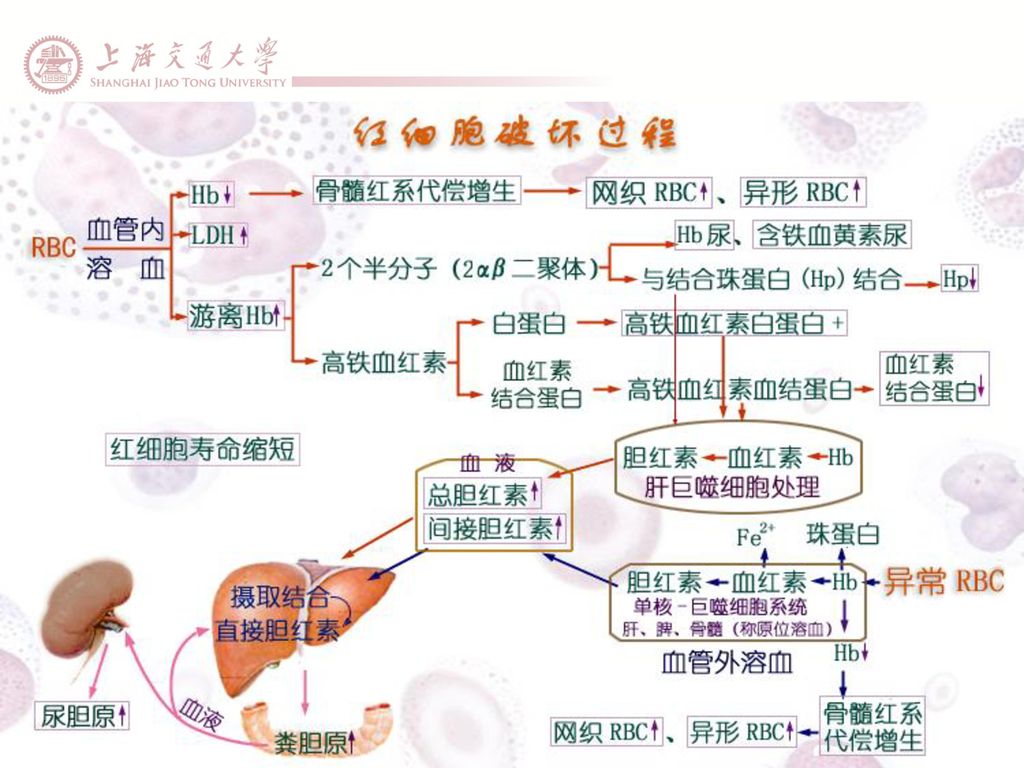
红细胞破坏增加的检查 胆红素代谢(非结合胆红素升高) 尿分析(尿 胆原升高) 血清结合珠蛋白(降低) 血清 游离 血红蛋白(升高) 尿血红蛋白(阳性) 乳酸脱氢酶(升高) 外周血涂片(破碎和畸形红细胞升高)
 尿分析(尿 胆原升高) 血清结合珠蛋白(降低) 血清 游离 血红蛋白(升高) 尿血红蛋白(阳性) 乳酸脱氢酶(升高) 外周血涂片(破碎和畸形红细胞升高)")
35
红细胞生成代偿增生的检查 网织红细胞计数(升高) 外周血涂片(出现有核红细胞) 骨髓检查(红系造血增生) 红细胞肌酸(升高)
 外周血涂片(出现有核红细胞) 骨髓检查(红系造血增生) 红细胞肌酸(升高)")
37
治疗原则 去除病因 糖皮质激素和其他免疫抑制剂 输血或成分输血 脾切除术 其他治疗

38
阵发性睡眠性血红蛋白尿 (PNH)
")
39
PNH的性质 获得性造血干细胞良性克隆性疾病 异常克隆不具有自主无限扩增特性 部分病例可以自愈

40
病因及发病机制 X染色体上PIG-A基因(Xp22.1)突变 导致糖化肌醇磷脂(GPI)锚合成障碍 己报道有100多种基因突变
具有异质性,可累及多个编码区及剪接点 一种异常细胞可有二种突变,同一患者可有一个以上异常克隆

41
GPI锚生物合成 (内质网中合成) N-乙酰葡萄糖胺 磷脂肌醇(PI) 脂质 核心结构 PI-葡糖胺 甘露糖 3-磷酸乙醇胺
 N-乙酰葡萄糖胺 磷脂肌醇(PI) 脂质 核心结构 PI-葡糖胺 甘露糖 3-磷酸乙醇胺")
42
GPI锚连接蛋白 补体调接蛋白 衰变加速因子(DAF或CD55)、膜攻击复合物抑制因子(MACIF或MIRL或CD59)、补体C8结合蛋白(HRF或C8bp)、膜辅助蛋白(MCP)。 粘附分子 淋巴细胞功能相关抗原-3(LFA-3或CD58)、 Blast-1/ CD48、CD67、CD66。
、 Blast-1/ CD48、CD67、CD66。")
43
GPI锚连接蛋白 酶类 红细胞乙酰胆碱脂酶 ( AchE )、中性粒细胞碱性磷酶酸酶 ( NAP )、5’外核酸酶 ( CD 73 )。
受体类 中性粒细胞III型Fc受体 ( FcRIIIb 或CD16 )、单核细胞分化抗原 ( CD14 )、尿激酶型纤溶酶原激活物受体 ( u-PAR )。 血型抗原 CD55 携带的Comer抗原、AchE携带的 Yt抗原。
、单核细胞分化抗原 ( CD14 )、尿激酶型纤溶酶原激活物受体 ( u-PAR )。 血型抗原. CD55 携带的Comer抗原、AchE携带的 Yt抗原。")
44
补体激活途径 CD55 CD59

45
PNH异常细胞克隆的维持和扩增 单独 PIG-A 基因突变不是发病唯一原因。 异常克隆并无增殖优势,正常造血细胞受到抑制时才得以扩增。

46
不同细胞群体外培养 细胞群 集落形成能力 CD34+ CD59+(正常) 正常 CD34+ CD59+(PNH) 显著减少
细胞群 集落形成能力 CD34+ CD59+(正常) 正常 CD34+ CD59+(PNH) 显著减少 CD34+CD59- (PNH) 减少
 正常. CD34+ CD59+(PNH) 显著减少. CD34+CD59- (PNH) 减少.")
47
PNH 异常细胞具有抗凋亡能力。 PNH患者具有GPI锚连蛋白的正常细胞易被T细胞识别而被杀伤,缺乏GPI锚连蛋白的造血细胞则可逃逸 。

48
PNH的临床表现 血红蛋白尿发作(血红蛋白管型,尿隐血阳性,含铁血黄素尿阳性) 全血细胞减少(网织红细胞轻中度增高)
血栓形成(下肢静脉性静脉炎、视网膜静脉血栓栓塞、脑血栓、Budd-Chiari综合征) 肾脏表现(主要为蛋白尿)
 肾脏表现(主要为蛋白尿)")
49
PNH和AA关系 相当密切,可以相互转化且并存 AA→PNH多(15%),PNH→AA少 20%`~30% PNH可伴骨髓再障
AA(ATG治疗后)→PNH 10~31% AA-PNH综合征 16.5%(我国) 50%AA外周血或骨髓可检出PNH克隆 AA→PNH 生存率要高于PNH→AA MDS-RA 如捡出PNH克隆者预后好,且ATG治疗有效。 PNH可视为对AA和MDS的“自然治疗”
→PNH 10~31% AA-PNH综合征 16.5%(我国) 50%AA外周血或骨髓可检出PNH克隆. AA→PNH 生存率要高于PNH→AA. MDS-RA 如捡出PNH克隆者预后好,且ATG治疗有效。 PNH可视为对AA和MDS的 自然治疗")
50
PNH的诊断 补体溶血实验 流式细胞术 嗜水气单胞菌毒素溶血实验

51
补体溶血试验 Ham试验阳性率79 %, 但特异性高。 糖水试验阳性率88 %,但易出现假阳性。 蛇毒因子溶血试验阳性率81%。
补体溶血敏感试验可将PNH红细胞分I、II、III型,临床溶血程度主要取决 III型。我国以I+II+III最多,I+III次之。

52
流式细胞术 直接定量测定,敏感性、特异性最高,Ham试验检出1%PNH细胞,流式可检出0.1%PNH细胞。
PNH克隆累及造血細胞次序为粒细胞→单核、红细胞→淋巴细胞,骨髓早于外周血,网红略早于红细胞。粒细胞CD59有早期诊断价值且受输血影响少。CD59敏感度高于CD55。 宜测定2种以上膜蛋白缺失,以排除遗传缺陷。

53
嗜水气单胞菌(HEC)毒素溶血试验 原理:毒素与GPI蛋白连接,破溶正常细胞,毒素作用后细胞留存率与CD59-率一致。
灵敏、特异、简便、价廉。

54
PNH的治疗 病因治疗 对症治疗:输血、补充造血因子、激素类药物、抗凝治疗….

55
造血干细胞移植治疗PNH 异基因骨髓移植是唯一能治愈本病方法 指征:PNH合併骨髓增生低下,且反复发生严重血管栓塞并发症者。

56
Eculizumab 人源性C5单抗,可抑制C5转化酶,阻止MAC形成 安全有效控制溶血的措施

57
注意点:ATG和ALG作为抗体与人血细胞表面抗原结合形成免疫复合物易激活补体 。
免疫抑制剂 ( ATG、ALG、CsA) 适用低增生性PNH 原理: 不论PNH或AA都有毒性T淋巴细胞克隆扩增,但不是针对锚蛋白。 注意点:ATG和ALG作为抗体与人血细胞表面抗原结合形成免疫复合物易激活补体 。
 适用低增生性PNH. 原理: 不论PNH或AA都有毒性T淋巴细胞克隆扩增,但不是针对锚蛋白。 注意点:ATG和ALG作为抗体与人血细胞表面抗原结合形成免疫复合物易激活补体 。")
58
基因治疗 Nishimura J 等(2001)以逆转录病毒为载体,将PIG-A基因转入缺失细胞,使其恢复GPI锚连蛋白表达。
亦可转入外周血CD34 +细胞。 是否会加重骨髓衰竭?

59
自身免疫性溶血性贫血 (AIHA)
")
60
抗体分类 自免溶贫抗体: 温抗体型(最佳温度37℃)(80.3%): 不完全温性抗体(IgG,IgM,IgA,混合型) 温性自身溶血素
冷抗体(最佳温度0-5℃)(19.7%) 冷凝集素综合症——冷凝激素性IgM 冷热抗体(Donath-Landsteiner)
(19.7%) 冷凝集素综合症——冷凝激素性IgM. 冷热抗体(Donath-Landsteiner)")
61
病因 原发性 继发性 淋巴增殖性肿瘤(CLL、淋巴瘤、骨髓瘤) 结缔组织疾病(SLE、类风关、硬皮病) 感染性疾病(细菌、病毒、支原体)
非淋巴系肿瘤(卵巢囊肿、肝癌) 药物(青霉素类、奎尼丁、甲基多巴、嘌呤核苷类似药fludarabine/cladribine)
 药物(青霉素类、奎尼丁、甲基多巴、嘌呤核苷类似药fludarabine/cladribine)")
62
发病机制 自身免疫耐受异常——DC调控异常、T和B细胞免疫耐受异常
免疫调节异常——包括Th1/Th2细胞组成的细胞核体液免疫调节“枢纽”以及“调节性T细胞” 抗体后调节异常 基因易感性——早年动物研究显示,存在2个优势即Aial和Aia2;新近发现7号染色体上的Aia3和1号染色体上的Nba2基因与发病有关。人类Fas基因缺失,可使自身抗体明显增加而导致AIHA。CTL相关抗原4(CTLA4)的第49外显子A→T突变与AIHA有明显相关性
的第49外显子A→T突变与AIHA有明显相关性.")
64
诊断 温抗体型AIHA: 近4月内无输血或特殊药物服用史 直接抗人球蛋白试验阳性 临床表现和实验室检查 抗人球蛋白试验阴性的AIHA:
临床表现较符合,肾上腺皮质激素或脾切除治疗有效 除外其他溶血性贫血 (冷抗体型AIHA各自临床表现结合相应的实验室检查,可作出诊断)
")
65
抗人球蛋白试验(Coombs试验) 实验原理:
由于红细胞表面带有负电荷,使红细胞相互排斥,而保持一定剧烈。不完全抗体如IgG可结合在红细胞膜上,称致敏红细胞。由于不完全抗体分子短,在盐水介质中不发生可见的凝集现象。 直接法DAT:若加入抗人球蛋白血清,其可特异性与红细胞表面不完全抗体分子结合(称抗体搭桥),使致敏红细胞能相互连接,出现可见的凝集现象。 间接法IAT:将正常Rh+O型红细胞与患者血清混合孵育,若血清中存在不完全抗体,可使红细胞致敏,再加入抗人球蛋白血清,观察凝集现象。
,使致敏红细胞能相互连接,出现可见的凝集现象。 间接法IAT:将正常Rh+O型红细胞与患者血清混合孵育,若血清中存在不完全抗体,可使红细胞致敏,再加入抗人球蛋白血清,观察凝集现象。")
67
根据特异性单价抗血清分为三种亚型: ——抗IgG和补体C3型,占67%,红细胞破坏最重,贫血最重 ——单独抗IgG占20%,溶血中等 ——单独抗补体C3型占13%,溶血最轻 ——抗IgA和抗IgM型均罕见

68
抗球蛋白试验假阴性 抗球蛋白试验阳性必须每个红细胞上至少有 个IgG分子或60-115个补体C3分子,低于此数,则常规DAT阴性。 用敏感的检测方法如生物素亲和系统和系统-抗球蛋白试验(BAS-Coombs)、亲和素生物素化酶复合物酶联免疫分析(ABC-ELISA)、抗IgG消耗、免疫荧光法测定、流式细胞术检测红细胞结合IgG等可检出抗红细胞抗体。 真正Coombs阴性的自免溶贫极少。
、亲和素生物素化酶复合物酶联免疫分析(ABC-ELISA)、抗IgG消耗、免疫荧光法测定、流式细胞术检测红细胞结合IgG等可检出抗红细胞抗体。 真正Coombs阴性的自免溶贫极少。")
69
抗球蛋白试验假阳性 有血块微粒 用硅胶管盛血 抽静脉血混有低离子强度溶液 高丙种球蛋白血症
药物如头孢菌素可致血浆蛋白非特异性吸附于红细胞表面 DAT阴性而IAT阳性可能系同种免疫抗体所致,与输血或妊娠有关,而不是自身免疫所致。

70
AIHA——溶血危象 贫血突然加重,黄疸加深 血管外溶血尿色呈浓茶样,血管内溶血则有血红蛋白尿,尿色呈葡萄酒色或酱油色 网织红细胞明显增高
脾脏增大 一般白细胞及血小板正常 骨髓为增生性贫血象

71
AIHA——再生障碍危象 贫血突然加重,甚至可出血,黄疸不加深 网织红细胞减低,甚至缺如 全血细胞减少,如为纯红再障危象则白细胞和血小板正常
骨髓象增生减低类似AA,如为纯红再障危象,则仅红系减少或缺乏,粒系和巨核系正常

72
治疗 积极寻找病因,治疗原发病 肾上腺糖皮质激素: 缓解率60-80%,约20-30%可达持续缓解 发生作用时间平均7天
主要机制:抑制淋巴细胞的抗体形成;干扰Ag-Ab反应,改变Ab对RBC抗原的亲和力;改变单核-巨噬系统对有Ab包裹细胞的清除作用,尤其是可明显抑制IgG敏感细胞的清除 大剂量静脉丙种球蛋白: 疗效——不及ITP,甚至偶尔反可加重溶血。文献报道,仅1/3病例取得暂时缓解 用药限制——仅限于其它疗法未能奏效而患者又必须在极短时间内取得缓解者 脾切除: 脾脏是抗体的生成器官,又是致敏红细胞的主要破坏场所 适应症:肾上腺皮质激素无效,或需较大剂量才能维持缓解,或因激素副作用明显 输血: 适应症:爆发性AIHA;溶血危象;极重度贫血短期内可能危及生命者

73
其它治疗 治疗进展——单克隆抗体: 美罗华(Rituximab)是基因工程技术合成的人鼠嵌合型抗CD20的单克隆抗体
Cammpath-1H是人源化的针对CD52的单克隆抗体,IgG1型 CD52表达在人;淋巴细胞和单核细胞上,其他血细胞上缺乏,单克隆抗体结合CD52+细胞后通过补体依赖的或抗体依赖的细胞毒作用,或通过细胞凋亡的机制清除CD52+的细胞,它可以长时间、严重的抑制外周血中的T、B细胞。 毒副作用与美罗华类似,尤其是停用该药后免疫抑制作用还能够持续一段时间,在此期间很容易发生感染 Eculizumab是补体C5的人源化的单克隆抗体,其Fc段几乎没有功能,与补体C5的亲和力高 与C5结合后能够抑制C5的裂解,抑制炎症介质C5a的释放和C5b-C9复合物的形成 保留了补体激活过程中的早期成分,因而补体对微生物的调理和清除免疫复合物的作用不受影响 人工合成肽段竞争结合自身抗体 细胞因子治疗 环孢素、双磷酸盐

74
End Thanks




. 免疫缺陷病 (Immunodeficiency diseade,IDD) : 由免疫系统中任何一个成分在发生、发 育和成熟过程中的缺失或功能不全而导致免 疫功能障碍所引起的疾病。 免疫缺陷病分为 : 先天性 />")
 特发性血小板减少性紫癜 (idiopathic thrombocytopenic purpura,ITP) 又称 免疫性血小板减少性紫癜是一种自 身免疫性出血性疾病,其特点是机.>")
 毛细血管采血 ( 二 ) 静脉采血 ( 三 ) 常用抗凝剂 1. 肝素 2. 柠檬酸钠.>")

 、 即向髓系细胞和淋巴系细胞分化。>")
. 二. 白细胞计数和 白细胞分类计数 白细胞起源、发育阶段 干细胞 祖细胞 前体细胞 成熟细胞.>")


 由于各种原因使红细胞寿命缩短,破坏 加剧,超过骨髓造血代偿能力所致。 由于各种原因使红细胞寿命缩短,破坏 加剧,超过骨髓造血代偿能力所致。 血涂片可为诊断提供重要线索,可见到 不同形态的红细胞和受损伤的表现。 血涂片可为诊断提供重要线索,可见到.>")
>")


 第三节 贫血的实验诊断 Laboratory Diagnosis of Anemia.>")


 魏彦刚 菏泽医学专科学校.>")

毛细血管采血 (二)静脉采血 (三)常用抗凝剂 1.肝素 2.柠檬酸钠.>")

