Download presentation
Presentation is loading. Please wait.

1
PCR技术及其应用 朱德裕 2013年11月1日

2
主要内容 一、PCR技术简史 二、PCR的基本原理 三、PCR的反应体系和方法 四、PCR的类型和应用

3
PCR技术简史 什么是PCR? 多聚酶链式反应(polymerase chain reaction)简称PCR技术,是一种体外扩增特异DNA片段的技术。 PCR技术操作简便,可在短时间获得数百万个特异的目的DNA序列的拷贝。80年代中期PCR技术的发明,引起了生物技术发展的一次革命,目前已被广泛应用于与分子生物学相关的各个领域。

4
DNA体内的半保留复制 DNA 聚合酶

5
核酸体外扩增的设想 1971年,哈尔.戈宾.科拉纳 (Har Gobind Khorana)首先提出:经过DNA变性,与合适引物杂交,用DNA聚合酶延伸引物,并不断重复该过程便可克隆tRNA基因。 但由于测序和引物合成的困难,以及70年代基因工程技术的发明使克隆基因成为可能,所以,Khorana的设想被人们遗忘了…… Har Gobind Khorana(1922-) 1968年获诺贝尔生理学或医学奖,因阐明遗传密码及其在蛋白质合成中的作用,
 1968年获诺贝尔生理学或医学奖,因阐明遗传密码及其在蛋白质合成中的作用,")
6
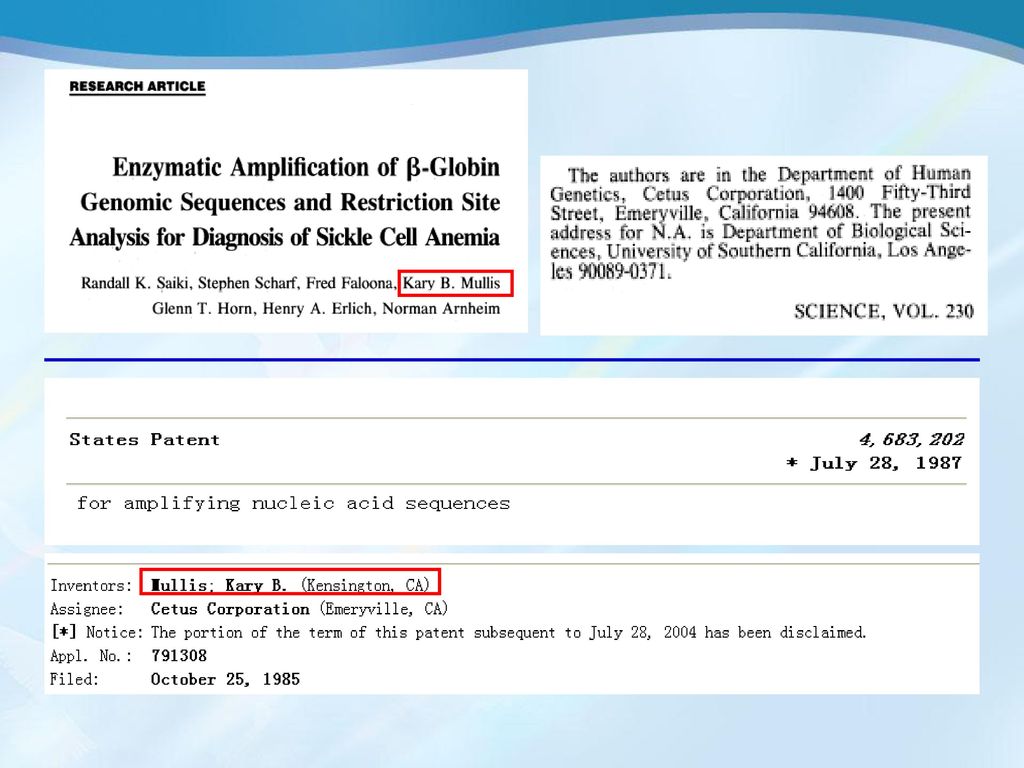
聚合酶链反应PCR的发明 1985年,美国PE-Cetus公司的Mullis等人发明了聚合酶链反应(PCR)。
基本原理是在试管中模拟细胞内的DNA复制。 耐热DNA聚合酶的应用使得PCR能高效率的进行,随后PE-Cetus公司推出了第一台PCR自动化热循环仪。 1989年美国《Science》杂志列PCR 为十余项重大科学发明之首,比喻1989年为PCR爆炸年。 1993年,Mullis等因此项技术获诺贝尔化学奖。 Kary B. Mullis (1944- )
")
7
Kary B. Mullis 是一位生化学家,又是一名冲浪爱好者。 1972在加州大学柏克莱分校获得博士学位,专业是有机合成。读博士期间,突然有天想出了某个解释宇宙大爆炸的理论,并写了出来投稿到Nature,居然被刊登了!他也因此拿到了博士学位。 1979年,Mullis进了一家叫Cetus的私人生物技术公司任职。 Cetus聘用Mullis,是看重他有机化学合成的专长,负责合成寡聚核苷酸,以供实验所用。Mullis花很多时间玩当时刚刚流行的个人电脑,也不时提出一些古怪的想法,其中大部分都是错的。 有段时间Mullis想使用染色的方法鉴定一个突变的基因(这是一个失败的想法,但却把他引向DNA的扩增)。一直在做突变检测工作导致Mullis想到细胞内DNA复制的反应。碰巧,Mullis那时一直沉迷于计算机如何处理对数功能,这导致他将两个事情联系在一起,从而产生了指数扩增DNA的构想。Mullis意识到,小的东西如果持续加倍,则会增加得很快。例如,将一个特定的DNA片段加倍30次,则会产生230拷贝的,这么多拷贝的DNA足以让研究者进行相关的科学研究。
。一直在做突变检测工作导致Mullis想到细胞内DNA复制的反应。碰巧,Mullis那时一直沉迷于计算机如何处理对数功能,这导致他将两个事情联系在一起,从而产生了指数扩增DNA的构想。Mullis意识到,小的东西如果持续加倍,则会增加得很快。例如,将一个特定的DNA片段加倍30次,则会产生230拷贝的,这么多拷贝的DNA足以让研究者进行相关的科学研究。")
8
故事发生在1983年 的春夏之交

9
那是一个星期五的晚上,Mullis开着银色的本田Civic,带着女友前往乡间的小屋度周末。在高速公路上开着车,瞬间他感觉两排路灯就是DNA的两条链,自己的车和对面开来的车象是DNA聚合酶,面对面地合成着DNA。于是他停下了车,叫醒了正在熟睡的女友,激动地解释起他的想法,即用两个引物(而不是一个引物)去扩增模板DNA 的想法。 引物1 引物2 DNA酶 Mullis原以为这样简单的想法,应该有人提出过,但搜索文献后却发现没有。

10
在“猛然顿悟”之后的3~5个月间,Mullis并没有任何行动。同年8月,Mullis首次在公司里正式作了有关PCR原理的报告,听者反应冷淡。一来,大家已经习惯了他的胡思乱想;再者,多数人的想法是,这个原理太简单了,如果可行的话,一定早有人做过,否则,一定有什么不可行之处,但也没有人明确说得出来,为什么不可行。 从1983年9月起,Mullis陆续进行了一些实验,由于Mullis以前没有接受过分子生物学的训练,公司派了技术员协助,前后一共有三位。这些人在PCR的发展上,发挥了重要的作用。1984年11月,Mullis的技术员首次取得可信的结果,证明了PCR的可行。于是在1985年初,公司决定又派上了日裔技术员才木 (Randall Saiki)。 到了1985年春天,Cetus递出了第一个专利申请。他们决定撰写两篇论文,一篇关于PCR的原理,由Mullis执笔先行发表,第二篇则集中在PCR的应用上,以Saiki的实验结果为主,随后推出。结果整个夏天,Mullis都在玩电脑,一再拖延论文的写作。到9月下旬有关PCR应用的文章写好寄出时,Mullis还没有动静。因此,第一篇提到PCR这个方法的论文,于1985年12月20日发表在Science上,Saiki为第一作者,Mullis则排第四。 直到12月,Mullis才将论文写好,由于太晚,投给Nature,Science,结果都被拒。 Mullis的文章两度被拒以后,公司里有人建议投给Methods of Enzymology,文章终于得到发表,只不过整整晚了一年,到1987年初才问世。
。 到了1985年春天,Cetus递出了第一个专利申请。他们决定撰写两篇论文,一篇关于PCR的原理,由Mullis执笔先行发表,第二篇则集中在PCR的应用上,以Saiki的实验结果为主,随后推出。结果整个夏天,Mullis都在玩电脑,一再拖延论文的写作。到9月下旬有关PCR应用的文章写好寄出时,Mullis还没有动静。因此,第一篇提到PCR这个方法的论文,于1985年12月20日发表在Science上,Saiki为第一作者,Mullis则排第四。 直到12月,Mullis才将论文写好,由于太晚,投给Nature,Science,结果都被拒。 Mullis的文章两度被拒以后,公司里有人建议投给Methods of Enzymology,文章终于得到发表,只不过整整晚了一年,到1987年初才问世。")
12
1985年10月25日申请了PCR的专利,1987年7月28日批准(专利号4,683,202 ),Mullis是第一发明人。
Cetus的主管(他们无意争功)向冷泉港实验室的Watson推荐,让Mullis在1986年5月举行的“人类分子生物学”专题研讨会中,报告PCR的原理及实际应用结果。 这是Mullis生平第一次受邀演讲,分子生物学界许多著名学者也都在场。结果Mullis表现不错,建立了往后人们的印象:PCR是他一手发明的。冷泉港专题研讨会的专刊于1986年底出版,Mullis挂了头名。 自此,PCR之名及其强大的应用性就广为人知了。然而,将PCR变成真正成熟技术的“临门一脚”,则是耐高温DNA聚合酶的引进。 PCR传奇--一个生物技术的故事。 保罗·拉比诺著,朱玉贤译。上海科技教育出版社, 1998。
向冷泉港实验室的Watson推荐,让Mullis在1986年5月举行的 人类分子生物学 专题研讨会中,报告PCR的原理及实际应用结果。 这是Mullis生平第一次受邀演讲,分子生物学界许多著名学者也都在场。结果Mullis表现不错,建立了往后人们的印象:PCR是他一手发明的。冷泉港专题研讨会的专刊于1986年底出版,Mullis挂了头名。 自此,PCR之名及其强大的应用性就广为人知了。然而,将PCR变成真正成熟技术的 临门一脚 ,则是耐高温DNA聚合酶的引进。 PCR传奇--一个生物技术的故事。 保罗·拉比诺著,朱玉贤译。上海科技教育出版社, 1998。")
13
1986年春,Mullis首次提出使用耐高温酶的想法。经过文献搜索,找到了两篇有关文献,较早的一篇是在美国做的,另一篇则是俄罗斯科学家的成果。
第一篇报道分离耐高温DNA聚合酶的工作,是一位来自台湾的钱嘉韵。她的导师对一种在黄石公园的温泉里发现的嗜热菌(Thermus aquaticus,Taq)感到好奇,就让钱嘉韵及另一位美国学生以该细菌为论文研究的题目。钱嘉韵成功地从该细菌分离出耐高温的Taq DNA聚合酶,并将研究成果发表在1976年的Journal of Bacteriology上。 美国黄石国家公园(yellowstone National Park ) Thermus aquaticus
感到好奇,就让钱嘉韵及另一位美国学生以该细菌为论文研究的题目。钱嘉韵成功地从该细菌分离出耐高温的Taq DNA聚合酶,并将研究成果发表在1976年的Journal of Bacteriology上。 美国黄石国家公园(yellowstone National Park ) Thermus aquaticus.")
14
Mullis虽然提出将Taq DNA聚合酶应用到PCR的建议,但当时并没有现成的酶可用,他得想办法自己分离。Cetus有全套分离蛋白质的设备,也有人愿意指导,但Mullis是个拖拉成性的人。等了几个月后,公司其他人只有自己动手,按着先前钱嘉韵等人发表的步骤,三个星期就纯化出Taq DNA聚合酶。1986年6月,Saiki首度将其应用于PCR,效果就好得惊人,可以说是一炮打响。Taq DNA聚合酶不但大大简化了PCR工作,同时其专一性及活性都比之前使用的大肠杆菌DNA酶更强,背景杂带也几乎都消除了。自此,PCR大获成功。 1988年,第一台PCR仪问世; 1991,Cetus公司以3亿美元的转让费将PCR相关专利转让给瑞士Hoffman LaRoche公司,并在此后的经营中又获得了数亿美元的分红。 由于Mullis的个性以及自身的缺点,穆里斯于1986年9月离开了Cetus ,同时也离开了科研。 Cetus给了他五个月的薪水及一万美元奖金,但按产业惯例,PCR的专利权属于Cetus公司。

15
Mullis于1993年获得诺贝尔化学奖。

16
二、PCR的基本原理 类似于DNA的天然复制过程,其特异性依赖于与靶序列两端互补的寡核苷酸引物。PCR由变性--退火--延伸三个基本反应步骤构成: ①变性(denature):模板DNA经加热至95℃左右一定时间后,使模板DNA双链或经PCR扩增形成的双链DNA解离,使之成为单链,以便它与引物结合,为下轮反应作准备。 95℃ 模板DNA
:模板DNA经加热至95℃左右一定时间后,使模板DNA双链或经PCR扩增形成的双链DNA解离,使之成为单链,以便它与引物结合,为下轮反应作准备。 95℃ 模板DNA.")
17
②退火(annealing)(复性):模板DNA经加热变性成单链后,将温度降至引物的Tm值左右或以下(55℃左右),引物与模板DNA单链的互补序列配对结合,形成杂交链。
50-55℃ 引物1 DNA引物 引物2

18
③延伸(extension):DNA模板--引物结合物在Taq DNA聚合酶的作用下,以dNTP为反应原料,靶序列为模板,按碱基配对与半保留复制原理,合成一条新的与模板DNA 链互补的半保留复制链。
72℃ Taq酶 引物1 DNA引物 引物2 Taq酶

19
以上三步为一个循环,每一循环的产物作为下一个循环的模板, 25~30 次循环后,大约2~3小时后,模板DNA的含量可以扩大100万倍以上。

20
PCR的基本原理示意图 1 2 3 1 2 3 4 5 22 55 72 94 时间(min) 温度 (℃) 高温变性 低温退火 适温延伸
重复1~3步 25~30轮 形成2条单链 DNA变性 目的DNA片段 扩增100万倍以上 子链延伸 DNA加倍 DNA单链 与引物复性 DNA双螺旋

21
PCR 动画 过 程 变 性 引 物 退 火 DNA 复制 1st cycle 2nd cycle 3rd cycle

22
PCR的特点 灵敏度高 简便、快速 对标本的纯度要求低 皮克(pg=10-12)量级扩增到微克(ug=10-6)水平
能从100万个细胞中检出一个靶细胞 病毒检测的灵敏度可达3个RFU 细菌检测的最小检出率为3个细菌 简便、快速 一次性加好反应液,2~4 小时完成扩增 扩增产物一般用电泳分析 对标本的纯度要求低 血液、体腔液、洗嗽液、毛发、细胞、活组织等组织的粗提DNA

23
琼脂糖凝胶电泳 PCR 3-4 小时 最终产物 紫外光观察

24
三、PCR反应体系与反应条件 1、标准的PCR反应体系 (50~100ul)
10×缓冲液 /10体积 4种dNTP混合物 各200umol/L 两条引物 各1u mol/L 模板DNA ~2ug Taq DNA聚合酶 ~ 5u Mg2+ mmol/L 双蒸水 至50~100ul

25
PCR引物设计 PCR反应中有两条引物,即5′端引物和3′引物。设计引物时以一条DNA单链为基准(常以信息链为基准),5′端引物与位于待扩增片段5′端上游的一小段DNA序列相同;3′端引物与位于待扩增片段3′端的一小段DNA序列互补。 基准 5′ ATAAGCCCGAAAGGTTCGTTGATC CTAGATAGGTTGCCGATCCGTAAA 3′ tccaacggctaggcattt 5’ 5’ ataagcccgaaaggttcg 3′ TATTCGGGCTTTCCAAGCAACTAG GATCTATCCAACGGCTAGGCATTT 5′

26
④两个引物之间不应存在互补序列,尤其是避免3 ′端的互补重叠。
引物设计的基本原则: ①引物长度:15-30bp,常用为20bp左右。 ②引物碱基:G+C含量以40-60%为宜,G+C太少扩增效果不佳,G+C 过多易出现非特异条带。ATGC最好随机分布,避免5个以上的嘌呤或嘧啶核苷酸的成串排列。 ③引物内部不应出现互补序列。 ④两个引物之间不应存在互补序列,尤其是避免3 ′端的互补重叠。

27
⑤引物与非特异扩增区的序列的同源性不要超过70%,引物3′末端连续8个碱基在待扩增区以外不能有完全互补序列,否则易导致非特异性扩增。
⑥ 引物的5 ′端可以修饰。如附加限制酶位点,引入突变位点,用生物素、荧光物质、地高辛标记,加入其它短序列,包括起始密码子、终止密码子等。

28
引物设计软件 Primer Premier5.0 (自动搜索)* Oligo6 (引物评价) DNAsis Omiga DNAstar
Primer prim’er ( 如果从质粒里扩增DNA,我认为设计引物几乎不用考虑那么多,直接抄序列或互补序列就可以。

29
简介- Premier5.0的使用 第一步,运行软件,点击File,选择DNA sequence

30
第二步,复制序列后后用ctrl+V键将目的基因拷贝到栏内,后应加数个N以备后续设计时加酶切位点及保护碱基。

31
第三步,点击enzyme图标,将所选质粒上的多克隆酶切位点加入左栏

32
选中OK键,分析目的基因中所含的酶切位点,选酶切位点时就应从Non-culters 的这些酶中选取

33
第四步,点击primer图标,点S图标(即5‘端引物),软件默认引物长为25碱基。
,软件默认引物长为25碱基。")
34
点击edit primer,开始设计5’端引物。将鼠标点在设计框上就可以修改了,即可从3端从右向左删除一些碱基,也可在引物的5‘端加入选好的酶切位点并在其前再加上保护碱基,完成后点analyze,认为可以后点OK。

35
第五步,设计3’端引物,点击左上角A图标(反义链),用鼠标拉动滑块至基因末端,并将待选标签放在一定位置使所设引物到达目的基因末端。
,用鼠标拉动滑块至基因末端,并将待选标签放在一定位置使所设引物到达目的基因末端。")
36
点击EDIT PRIMERS图标,开始编辑3‘端引物,将酶切位点及几个保护碱基加在5‘端(记住酶切序列从右至左加入,即从从引物的5’端加),完成后点analyze,认为可以后点OK。
,完成后点analyze,认为可以后点OK。")
37
最后分析结果如图,反义链的FALSE PRIMING可以不考虑,RATING表示引物评分也可以不考虑,主要看Tm值正义链和反义链相差不应超过3度。GC含量不应超过60%。

38
此外,使用search功能,一般在设计RT-PCR检测的引物等可以使用。点击SEARCH图标,选择参数,一般选PCR primers---both—100至250个碱基,引物长短20+/-2,search parametere中的参数可以不选,为默认设置。点OK。

39
显示满足设计参数的候选结果,点OK.

40
分别点SENES、Anti-senes选择两个引物,RATING表示引物评分,最好选评分高的。整个一队引物评价同目的基因克隆的引物设计相同

41
模板 PCR的模板可以是DNA,也可以是RNA。

42
DNA聚合酶 Mullis最初使用的DNA聚合酶是大肠杆菌 DNA 聚合酶 I 的Klenow片段,其缺点是:
要重新添加。 ②容易发生模板和引物之间的碱基错配,其PCR产物特异性较差,合成的DNA片段不均一。 1988年初,改用T4 DNA聚合酶进行PCR,其扩增的DNA片段很均一,真实性也较高,只有所期望的一种DNA片段。但每循环一次,仍需加入新酶。

43
Taq酶 1988年Saiki 等从一株水生嗜热杆菌(thermus aquaticus) 中提取到一种耐热Taq DNA聚合酶。 此酶具有以下特点: ①耐高温,在70℃下反应2h后其残留活性大于原来的90%, 在93℃下反应2h后其残留活性是原来的60%,在95℃下反应 2h后其残留活性是原来的40%。 ②在热变性时不会被钝化,不必在每次扩增反应后再加新酶。 ③大大提高了扩增片段特异性和扩增效率,增加了扩增长度 (2.0Kb)。 此酶的发现使PCR广泛的被应用。
。 此酶的发现使PCR广泛的被应用。")
44
Taq酶的选择 高特异性:如QIAGEN公司的HotStar系列
高保真Taq酶:因其具有3”到5“核酸外切酶活性,错配率10-6,),如Stratagene的Pfu,NEB公司的Vent DNA聚合酶, QIAGEN公司的ProofStar DNA聚合酶,兼特异性与保真性于一体 超长片段扩增Taq酶;Clontech公司的Advantage Genomic系列混有TthDNA聚合酶,Proofreading酶和热启动抗体,对复杂模板可扩增10kb片段,简单模板可达40kb。 关于复杂模板的扩增 对于模板结构复杂,如GC含量高(>60%),有二级结构等,普通的Taq酶可能难以延伸下去,加入DMSO等助溶剂可帮助扩增顺利进行,但DMSO有毒,而且用量需要优化。QIAGEN公司的任何一种Taq酶都附有特制的Q-溶液,操作方便、安全,对复杂模板的扩增特别有效。
,如Stratagene的Pfu,NEB公司的Vent DNA聚合酶, QIAGEN公司的ProofStar DNA聚合酶,兼特异性与保真性于一体. 超长片段扩增Taq酶;Clontech公司的Advantage Genomic系列混有TthDNA聚合酶,Proofreading酶和热启动抗体,对复杂模板可扩增10kb片段,简单模板可达40kb。 关于复杂模板的扩增 对于模板结构复杂,如GC含量高(>60%),有二级结构等,普通的Taq酶可能难以延伸下去,加入DMSO等助溶剂可帮助扩增顺利进行,但DMSO有毒,而且用量需要优化。QIAGEN公司的任何一种Taq酶都附有特制的Q-溶液,操作方便、安全,对复杂模板的扩增特别有效。")
45
标准的PCR反应步骤设定 95℃ 15-30S 72℃,1kb/min PCR循环(22-30)
55℃,低于引物Tm值5-10 ℃左右,30s-55s 72℃,1kb/min 变性 退火 延伸 在Tm值允许范围内,选择较高的退火温度可大大减少引物和模板间的非特异性结合。 PCR循环(22-30) 反应的最后延伸和终止 72度进一步延伸5-10min,然后冷却至4度。
 反应的最后延伸和终止. 72度进一步延伸5-10min,然后冷却至4度。")
46
PCR扩增产物的分析 凝胶电泳分析 琼脂糖凝胶电泳: 通常应用1~2%的琼脂糖凝胶,供检测用。 聚丙烯酰胺凝胶电泳:6~10%聚丙烯酰胺凝胶电泳分离效果比琼脂糖好,条带比较集中,可用于科研及检测分析。 紫外线/灯下观察结果

47
讨论 PCR技术

48
四、PCR的类型和应用 研究 诊断 人类基因组工程 法医 肿瘤 其他…… 基因克隆,DNA测序,分析突变 细菌、病毒、寄生虫检测,诊断
犯罪现场标本分析 肿瘤 各种肿瘤检测 其他……

49
生物学领域几乎无处不用 基因、DNA片段的克隆 人工基因构建 DNA序列测定 基因定点突变 基因型(突变)检测, SNP分析,遗传背景分析
生物物种鉴定,系统进化研究 基因表达量研究(real-time PCR) / 基因表达谱研究( DNA chip, SAGE)
 / 基因表达谱研究( DNA chip, SAGE)")
50
医学领域 疾病基因检测 / 遗传病的产前诊断 致病病原体的检测 肿瘤治疗中癌基因的检测 会推广到大部分疾病治疗前的检测
DNA指纹、个体识别、亲子关系鉴别、法医物证 其他: 动、植物检疫(转基因动植物检测)
")
51
PCR相关的术语和产品层出不穷 TAS T-vector Multiplex PCR HotStart Taq Immuno PCR
Asymmetric PCR LP-PCR NASBA Recombinant PCR AFLP SSCP In situ PCR TaqMan/SYBR green T-vector HotStart Taq Reverse transcription PCR RAPD-PCR DDRT-PCR LM-PCR Inverse PCR Nested PCR Real-time PCR RACE Flow chip PCR

52
RT-PCR:反转录PCR(reverse transcriptase-PCR)
原理:RNA的反转录(RT)和cDNA的聚合酶链式扩增(PCR)相结合的技术。首先经逆转录酶的作用从RNA合成 cDNA,再以cDNA为模板,扩增合成目的片段。
和cDNA的聚合酶链式扩增(PCR)相结合的技术。首先经逆转录酶的作用从RNA合成 cDNA,再以cDNA为模板,扩增合成目的片段。")
53
特点: 作为模板的RNA可以是总RNA、mRNA或体外转录的RNA产物。无论使用何种RNA,关键是确保RNA中无RNA酶和基因组DNA的污染。用于反转录的引物可视实验的具体情况选择随机引物、Oligo dT 及基因特异性引物(GSP)中的一种。对于短的不具有发卡结构的真核细胞mRNA,三种都可。 应用:RT-PCR技术灵敏而且用途广泛,分析基因的转录水平,细胞中RNA病毒的含量,直接克隆特定基因的cDNA序列。

54
逆转录酶 逆转录酶是逆转录病毒RNA编码的,是多功能酶: ①具有RNA指导的DNA聚合酶活性,能和其它DNA聚合酶一样,沿5′ 3′方向合成DNA,并需要引物提供3′-OH ; ②具有RNA酶H(RNaseH)活性,能特异性水解RNA-DNA杂交体上的RNA; ③具有DNA指导的DNA聚合酶活性,以逆转录合成的单链DNA为模板合成互补DNA链。 逆转录酶对DNA没有3′ 5′外切酶活性,因此它没有校对功能,错误率相对较高。

55
RT-PCR的反应体系 逆转录体系 MgCl2 4μl(5m mol/L) Buffer 2 μl(1×) dNTP混合物 8 μl(1 μmol/L ) RNase抑制剂 0.5 μl(1u/ μl ) 反转录酶 1 μl(0.25u/ μl) 随机引物混合物 1 μl(2.5 μmol/L ) 总RNA 1 μl(1 μg) 无RNase的dH2O 至总体积20 μl 逆转录反应在420C进行1h 后,加热至950C5min,灭活逆转录酶。取2 μl反应产物,按DNA为模板的PCR反应步骤,进行扩增反应。
 随机引物混合物. 1 μl(2.5 μmol/L ) 总RNA. 1 μl(1 μg) 无RNase的dH2O. 至总体积20 μl. 逆转录反应在420C进行1h 后,加热至950C5min,灭活逆转录酶。取2 μl反应产物,按DNA为模板的PCR反应步骤,进行扩增反应。")
56
讨论 RNA提取与RT-PCR

57
一步法RT-PCR示意图 Stratagene一步法RT-PCR试剂盒是一个仅需一步反应就能简便、高通量地完成RT-PCR反应的高效系统。

58
从定性到定量的革命 聚合酶链式反应(PCR)可对特定核苷酸片断进行指数级的扩增 。在扩增反应结束之后,我们可以对扩增产物进行定量、定性的分析,但分析的都是 PCR 终产物。然而在许多情况下,我们所感兴趣的是未经 PCR 信号放大之前的起始模板量。例如我们想知道某一转基因动植物转基因的拷贝数或者某一特定基因在特定组织中的表达量。在这种需求下实时荧光定量PCR技术(Real time Quantitative PCR)应运而生。
可对特定核苷酸片断进行指数级的扩增 。在扩增反应结束之后,我们可以对扩增产物进行定量、定性的分析,但分析的都是 PCR 终产物。然而在许多情况下,我们所感兴趣的是未经 PCR 信号放大之前的起始模板量。例如我们想知道某一转基因动植物转基因的拷贝数或者某一特定基因在特定组织中的表达量。在这种需求下实时荧光定量PCR技术(Real time Quantitative PCR)应运而生。")
59
实时荧光定量PCR技术,是指在PCR反应体系中加入荧光基团,利用荧光信号的变化实时检测PCR扩增反应中每一个循环扩增产物量的变化,通过Ct值和标准曲线的关系对起始模板进行定量分析。
实时荧光定量PCR是目前确定样品中DNA(或cDNA) 拷贝数最敏感、最准确的方法。
 拷贝数最敏感、最准确的方法。")
60
Real Time PCR and Conventional PCR
VS 适用定性分析,不适合定量分析; PCR 产物的长度从100bp -数kb 无法对起始模板准确定量,无法对扩增反应实时检测 实时检测:每个循环都产出荧光信号 绝对定量,灵敏度更高 PCR产物的长度一般在 bps

61
荧光定量PCR原理--常用名词概念 扩增曲线 荧光阈值 Ct值

62
扩增曲线 Real Life 不同样品有不同的生成曲线 Theoretical 每个循环进行一次荧光信号的收集 Cycles
Log Target DNA Cycles 平台期 Rn(荧光强度) 每个循环进行一次荧光信号的收集 Lg-liner phase Baseline Cycle(循环数)
 每个循环进行一次荧光信号的收集. Lg-liner phase. Baseline. Cycle(循环数)")
63
荧光阈值 前15个循环信号作为荧光本底信号(baseline) 荧光阈值的缺省设置是3~15个循环的荧光信号的标准偏差的10倍
手动设置:大于荧光背景值和阴性对照的荧光最高值;进入指数期的最初阶段 真正的信号:荧光信号超过阈值 平台期 Rn(荧光强度) Lg-liner phase Threshold Baseline Cycle(循环数)
 Lg-liner phase. Threshold. Baseline. Cycle(循环数)")
64
Ct值 Ct值的定义: PCR扩增过程中,扩增产物的荧光信号达到设定的阈值时所经过的扩增循环次数 Rn(荧光强度) Ct value
Cycle(循环数)
")
65
Ct值的重现性 横轴:PCR反映循环数 纵轴:荧光信号量 Ct值的特点: 相同模板进行96次扩增,终点处产物量不恒定;Ct值极具重现性

66
Ct值与模板起始浓度的关系 模板起始浓度越高,Ct值越小 经数学证明,Ct值与模板DNA的起始拷贝数(dRn)成反比 。
成反比 。")
67
Ct值和起始模板量的对应关系 Template abundance in 6 samples. Low Ct, high amount
Cycle 23.8 23.7 23.6 Low Ct, high amount Practice with the animation so you understand the timing of this slide (if you haven’t used it before) - ask the audience to participate in this, it is a good way for you to determine the different comfort levels your audience has with the Ct concept. 35.6 35.3 35.2 High Ct low amount
 - ask the audience to participate in this, it is a good way for you to determine the different comfort levels your audience has with the Ct concept High Ct low amount.")
68
常用荧光标记方法 非特异性荧光标记 SYBR Green I 特异性荧光标记 TaqMan Probe

69
SYBR Green I染料法 SYBR Green I
SYBR Green I是一种非特异性结合于所有dsDNA双螺旋小沟区域的具有绿色激发波长的染料。 SYBR Green I

70
SYBR Green 能结合到双链DNA的小沟部位
退火和延伸时,形成双链DNA, SYBR Green 发荧光,在此阶段采集荧光信号。

71
SYBR Green I 染料法——作用机理 热 变 性 引物退火 延伸反应

72
SYBR green 的优点 SYBR green 的缺点 简便 可以使用已有的引物 普遍通用 可以检测所有的双链DNA,包括引物二聚体;
价格便宜 SYBR green 的缺点 容易与非特异性双链DNA结合,产生假阳性 需要化大力气优化反应条件,以消除非特异性扩增; 对引物特异性要求较高

73
引物设计原则 关键点: 设计合适引物,防止非特异性扩增! 长度:20-27nt G+C含量:45-55%
关键点: 设计合适引物,防止非特异性扩增! 引物设计原则 长度:20-27nt G+C含量:45-55% 碱基的随机分布:3’端不应超过3个连续的G或C 引物自身:不应存在互补序列 引物之间:不应有互补性,尤应避免3’端的互补重叠 引物的3’端:3’也不能有形成任何二级结构的可能 引物的特异性 推荐网站: RTPrimerDB ,实验证明有效的已设引物数据库

74
特异性荧光标记Taqman探针法 5′端标记有报告基团(Reporter, R) ,如FAM、VIC等
3′端标记有荧光淬灭基团 (Quencher, Q) 探针完整,R发射的荧光能量被Q基团吸收 ,无荧光 Taq酶有 5′→3′外切核酸酶活性,可水解探针, R与Q分开,发荧光 Taqman探针具有序列特异性,只结合到互补区 只要设计一条探针,先选择好探针,然后设计引物使其尽可能的靠近探针
 探针完整,R发射的荧光能量被Q基团吸收 ,无荧光. Taq酶有 5′→3′外切核酸酶活性,可水解探针, R与Q分开,发荧光. Taqman探针具有序列特异性,只结合到互补区. 只要设计一条探针,先选择好探针,然后设计引物使其尽可能的靠近探针.")
75
Taqman探针法——工作机理 每扩增一条DNA分子,释放一个荧光信号,可以在循环过程中任一点检测荧光
报告基团 每扩增一条DNA分子,释放一个荧光信号,可以在循环过程中任一点检测荧光 淬灭基团 反应体系中包括一对引物和一条探针,PCR中,Taq酶在链的延伸过程中遇到与模版结合的探针,其5’-3’外切酶活性的作用,将使探针的5’端的R被切断,加大了与Q的距离,从而使荧光恢复。一分子的产物生成就伴随着一分子的荧光信号的产生。随着扩增循环数的增加,释放出来的荧光基团不断积累。因此,Taqman探针检测的是积累荧光。

76
Taqman探针法——PCR体系的建立 1、引物、探针的设计:
探针Tm为68-70℃ ,比引物TM值高5-10℃(至少要5℃), <30 bp, 5’不能有G,G可能会淬灭荧光素,首先考虑探针的设计。 引物尽量靠近探针,扩增片段<400 bp,引物Tm为59-60℃ 2、反应参数的确定: 一般为:94 ℃,10-20S 60℃,30-60S(Taq酶5′→3′外切核酸酶活性在60℃ 最高),也可通过温度梯度优化退火温度 3、优化引物和探针浓度:获得最小Ct值,最大信号/背景比值 引物浓度: nM 探针浓度:50-300nM 4、其他与常规PCR相同
, <30 bp, 5’不能有G,G可能会淬灭荧光素,首先考虑探针的设计。 引物尽量靠近探针,扩增片段<400 bp,引物Tm为59-60℃ 2、反应参数的确定: 一般为:94 ℃,10-20S. 60℃,30-60S(Taq酶5′→3′外切核酸酶活性在60℃ 最高),也可通过温度梯度优化退火温度. 3、优化引物和探针浓度:获得最小Ct值,最大信号/背景比值. 引物浓度: nM. 探针浓度:50-300nM. 4、其他与常规PCR相同.")
77
Taqman探针法——优缺点 优 点 缺 点 高度特异性 重复性好 灵敏度高 可进行多重定量 只适合一个特定的目标 委托公司标记,价格较高
不易找到本底低的探针

78
TaqMan系统的一个例子 Titrate a template; 10, 1, 0.1, 0.01, 0.001 ng 10ng
HERE IS ANOTHER EXAMPLE SHOWING UNIFORM SPACING OF A SERIAL DILUTION, ROBUSTNESS IN REPLICATES, AND RANGE OF ASSAY Titrate a template; 10, 1, 0.1, 0.01, ng 78

79
讨论和比较 一般PCR与实时定量PCR的相同及不同

80
荧光定量PCR技术的应用 定性分析研究:杂合或纯合子鉴定,(SNPs)分析等
绝对定量研究:病毒和病原菌定量分析,基因拷贝数定量,GMO定量检测等 相对定量研究:mRNA表达量分析,siRNA效果确认, 差异显示结果验证等

81
利用TaqMan法检测血液中的HBV 已肝病人血液中HBV的绝对定量 方法:从血液中提取病毒DNA,扩增病毒基因,以TaqMan探针进行检测
设置对照:浓度为106、105 、104、103 的标准样品各一个,设阴性和空白对照 实验步骤:提取HBV DNA 设计特异引物 设计TaqMan探针并标记探针 扩增程序 结果:获取血液样品中HBV DNA的精确copy数

82
分析结果: HBV DNA的精确copy数为3.7X105
数据分析 分析结果: HBV DNA的精确copy数为3.7X105

83
多重PCR (Multiplex PCR) 一般PCR仅应用一对引物,通过PCR扩增产生一个核酸片段,主要用于单一致病因子等的鉴定。
多重PCR是在同一PCR反应体系里加上二对以上引物,同时扩增出多个核酸片段的PCR反应,其反应原理,反应试剂和 操作过程与一般PCR相同。 多重PCR的主要用于多种病原微生物的同时检测或鉴定和病原 微生物,某些遗传病及癌基因的分型鉴定。比如:①肝炎病毒的感染,在同一病人或同一供血者体内,有时存在多种肝炎病毒重叠感 染,有时是甲乙丙型肝炎病毒重叠;有时可能是甲乙型肝炎病毒重叠;有时是乙丙型肝炎病毒重叠.②肠道致病性细菌的检测,如伤寒,痢疾和霍乱,有时具有较相 同的肠道症状,有时痢疾霍乱同存一病人并同时发病.③性病的检测,如梅毒,淋病及艾滋病的诊断.④战伤细菌及生物战剂细菌的检测,如破伤风杆菌,产气荚膜 杆菌,炭疽杆菌,鼠疫杆菌等侦检.

84
多重PCR的特点有: ①高效性,在同一PCR反应管内同时检出多种病原微生物,或对有多个型别的目的基因进行分型,特别是用一滴血就可检测多种病原 体. ②系统性,多重PCR很适宜于成组病原体的检测,如肝炎病毒,肠道致病性细菌,无芽胞厌氧菌,及细菌战剂的同时侦检. ③经济简便 性,多种病原体在同一反应管内同时检出,将大大的节省时间,节省试剂,节约经费开支,为临床提供更多更准确的诊断信息.

85
高通量核酸扩增技术一直是大规模核酸样品检测分析的瓶颈。
多重PCR技术的难点在于多对引物的设计 多重引物的设计有几个原则: (a)各个引物对的Tm等参数尽量接近 (b)各个引物对应本本身就是一对优秀的单独的引物对,不要存在严重的二级结构比如发夹,dimers以及误引发。 (c)多重体系中的各个引物,应均按两两组合配对检查两个引物之间会不会存在严重的dimers,记住,是每条引物都要与其他引物配对检查。 高通量核酸扩增技术一直是大规模核酸样品检测分析的瓶颈。
各个引物对的Tm等参数尽量接近 (b)各个引物对应本本身就是一对优秀的单独的引物对,不要存在严重的二级结构比如发夹,dimers以及误引发。 (c)多重体系中的各个引物,应均按两两组合配对检查两个引物之间会不会存在严重的dimers,记住,是每条引物都要与其他引物配对检查。 高通量核酸扩增技术一直是大规模核酸样品检测分析的瓶颈。")
86
PCR还有很多改进的余地,而改进的诀窍可能就在多聚酶那里,因为PCR反应是人为的,而在生物体内,DNA的复制是有许多的酶和其它辅助蛋白参与。
创新的机会可能是去当第一个“把生米煮成熟饭”的人,也可能是去把熟饭“回锅”从新炒一番。

87
PCR仪

88
PCR仪的种类总体来说可以分为两大类: 普通PCR扩增仪和实时荧光定量PCR仪,普通的PCR扩增仪又衍生出带梯度PCR功能的梯度PCR仪、和带原位扩增功能的原位PCR仪等等。1996年由ABI公司首先推出将扩增和检测融为一体的实时荧光定量PCR仪,此后很多公司如Eppendorf、ROCHE、Corbett、MJ等等都先后推出不同款式的定量PCR仪。

90
实时定量PCR仪 7500型定量PCR仪 ViiA™ 7定量PCR仪

Similar presentations









DNA、RNA及蛋白质操作技术.>")










