Download presentation
1
第3章 化学平衡 Chemical Equilibrium 主讲教师:公瑞煜

3
本 章 要 点 (1) 化学反应的平衡条件 (2) 化学反应等温方程式和平衡常数 (3) 平衡常数表示法
(4) 平衡常数的测定和平衡转化率的计算 (5) 标准状态下反应的吉布斯能变化及化合物的标准生成自由能 (6) 温度对平衡常数的影响 (7) 其他因素对平衡的影响 (8) 反应的耦合
 平衡常数的测定和平衡转化率的计算. (5) 标准状态下反应的吉布斯能变化及化合物的标准生成自由能. (6) 温度对平衡常数的影响. (7) 其他因素对平衡的影响. (8) 反应的耦合.")
4
3.1 化学反应的平衡条件 化学反应体系 热力学基本方程 化学反应的方向与限度 为什么化学反应通常不能进行到底

5
3.1.1 化学反应体系 化学反应体系: 封闭的单相体系,不作非膨胀功,发生了一个化学反应,设为: aA + dD gG + hH
3.1.1 化学反应体系 化学反应体系: 封闭的单相体系,不作非膨胀功,发生了一个化学反应,设为: aA + dD gG + hH 各物质的变化量必须满足: 根据反应进度的定义,可以得到:

6
3.1.2 热力学基本方程 等温、等压条件下, 当 时:

7
这两个公式适用条件: (1)等温、等压、不作非膨胀功的一个化学反应; (2)反应过程中,各物质的化学势 保持不变。 公式(a)表示有限体系中发生微小的变化; 公式(b)表示在大量的体系中发生了反应进度等于1 mol的变化。这时各物质的浓度基本不变,化学势也保持不变。
等温、等压、不作非膨胀功的一个化学反应; (2)反应过程中,各物质的化学势 保持不变。 公式(a)表示有限体系中发生微小的变化; 公式(b)表示在大量的体系中发生了反应进度等于1 mol的变化。这时各物质的浓度基本不变,化学势也保持不变。")
8
3.1.3 化学反应的方向与限度 用 判断都是等效的。 反应自发地向右进行 反应自发地向左进行,不可能自发 向右进行 反应达到平衡

9
用 判断,这相当于 图上曲线的斜率,因为是微小变化,反应进度处于0~1 mol之间。
反应自发向右进行,趋向平衡 反应自发向左进行,趋向平衡 反应达到平衡

10
3.1.4 为什么化学反应通常不能进行到底? 严格讲,反应物与产物处于同一体系的反应都是可逆的,不能进行到底。
3.1.4 为什么化学反应通常不能进行到底? 严格讲,反应物与产物处于同一体系的反应都是可逆的,不能进行到底。 只有逆反应与正反应相比小到可以忽略不计的反应,可以粗略地认为可以进行到底。这主要是由于存在混合吉布斯自由能的缘故。

11
将反应 为例,在反应过程中吉布斯自由能随反应过程的变化如图所示。
R点,D和E未混合时吉布斯自由能之和; P点,D和E混合后吉布斯自由能之和; T点,反应达平衡时,所有物质的吉布斯自由能之总和,包括混合吉布斯自由能; S点,纯产物F的吉布斯自由能。

12
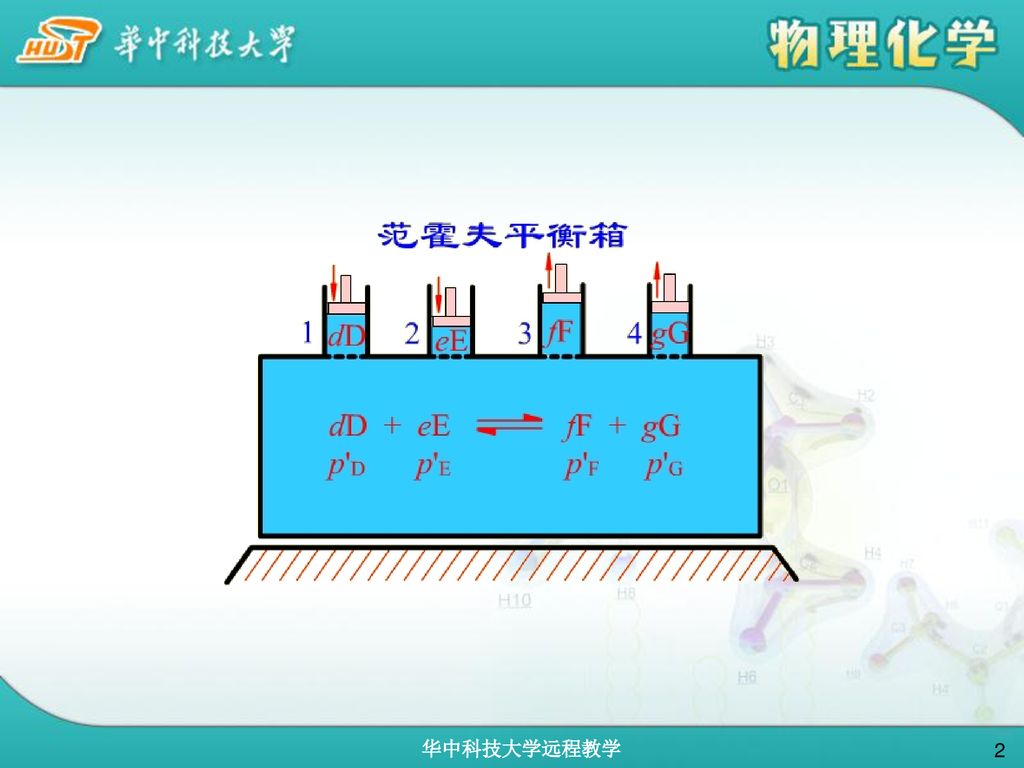
若要使反应进行到底,须在van’t Hoff 平衡箱中进行,防止反应物之间或反应物与产物之间的任何形式的混合,才可以使反应从R点直接到达S点。
G 最小值原理:当化学反应达平衡时,反应体系的G达最小值。

13
3.1.5 化学反应亲和势(affinity of chemical reaction)
1922年,比利时热力学专家德唐德(De donder)首先引进了化学反应亲和势的概念。他定义化学亲和势A为: 或 A是状态函数,体系的强度性质。用A判断化学反应的方向具有“势”的性质,即: A>0 反应正向进行 A<0 反应逆向进行 A=0 反应达平衡
首先引进了化学反应亲和势的概念。他定义化学亲和势A为: 或. A是状态函数,体系的强度性质。用A判断化学反应的方向具有 势 的性质,即: A>0 反应正向进行. A<0 反应逆向进行. A=0 反应达平衡.")
14
3.2 化学反应等温方程式和平衡常数 任何气体B化学势的表达式 化学反应等温方程式 热力学平衡常数 用化学反应等温式判断反应方向

15
3.2.1 任何气体B化学势的表达式: 式中fB为逸度,如果气体是理想气体,则fB=pB 。 将化学势表示式代入 的计算式,得:
将化学势表示式代入 的计算式,得: 称为化学反应标准摩尔Gibbs 自由能变化值( Standard change of molar Gibbs function of a reaction ) ,只是温度的函数。
 ,只是温度的函数。")
16
3.2.2 化学反应等温方程式 aA + dD gG + hH 有任意反应
3.2.2 化学反应等温方程式 aA + dD gG + hH 有任意反应 这就是化学反应等温方程式。 Qf称为“逸度商”,可以通过各物质的逸度求算。ΔrGmØ(T)值也可以通过多种方法计算,从而可得ΔrGm 的值。
值也可以通过多种方法计算,从而可得ΔrGm 的值。")
17
3.2.3 热力学平衡常数 当体系达到平衡,ΔrGm=0,则
3.2.3 热力学平衡常数 当体系达到平衡,ΔrGm=0,则 KfØ称为热力学平衡常数,它仅是温度的函数。在数值上等于平衡时的“逸度商”,是量纲为1的量,单位为1。因为它与标准化学势有关,所以又称为标准平衡常数。

18
3.2.4 用化学反应等温式判断反应方向 化学反应等温式也可表示为: 对理想气体 反应向右自发进行 反应向左自发进行 反应达平衡

19
3.3 平衡常数表示法 经验平衡常数 平衡常数与化学方程式的关系

20
3.3.1 平衡常数与化学方程式的关系 下标 m 表示反应进度为 1 mol 时的标准Gibbs自由能的变化值。显然,化学反应方程中计量系数呈倍数关系,ΔrGmØ(T)的值也呈倍数关系,而 KfØ 值则呈指数的关系。 例如: (1) (2)
 (2)")
21
3.3.2 经验平衡常数 反应达平衡时,用反应物和生成物的实际压力、摩尔分数或浓度代入计算,得到的平衡常数称为经验平衡常数,一般有单位。例如,对任意反应: aA + dD gG + hH 1. 用压力表示的经验平衡常数Kp 当 时, 的单位为1,无量纲。

22
2.用摩尔分数表示的平衡常数Kx 对理想气体,符合Dalton分压定律,pB=pxB

23
3.用物质的量浓度表示的平衡常数Kc 对理想气体,

24
4.液相反应用活度表示的平衡常数Ka 因为 ,则

25
例题: 右旋葡萄糖在乙醇水溶液中α型和β型的溶解度分别为0.11和0.27mol·dm-3,其无水固体在298K时的标准摩尔生成吉布斯函数分别为-902.9和 kJ.mol-1,试求298K时在上述溶液中α型和β型相互转化的Kø。 解: 对反应 α-C6H12O6(aq)= β- C6H12O6(aq) 关键:如何求乙醇水溶液中的ΔfGmø
= β- C6H12O6(aq) 关键:如何求乙醇水溶液中的ΔfGmø.")
26
确定标准态: 根据所给溶解度数据,取c=1mol·dm-3且仍遵守亨利定律pB= kcB的假想态为标准态。 设计如下途径: ΔG α-C6H12O6(s) α-C6H12O6(aq,c=cø) ΔG 2 ΔG 1 α-C6H12O6( aq,饱和溶液 c = 0.11mol·dm-3 ) ΔG = ΔG 1 + ΔG 2
 ΔG = ΔG 1 + ΔG 2.")
27
ΔG1=0 ΔG =ΔG2 = RTln(cø/0.11mol·dm-3)=5.47kJ·mol-1 ΔG = ΔfGmø (α,aq) - ΔfGmø (s) ΔfGmø (α,aq) = ΔfGmø(s) + ΔG = ( ) kJ·mol-1 = kJ·mol-1 同理可得: ΔfGmø (β,aq) = kJ·mol-1 ΔrGmø= kJ·mol-1 Kø= exp(- ΔrGmø/RT) = 1.27
 kJ·mol-1. = kJ·mol-1. 同理可得: ΔfGmø (β,aq) = kJ·mol-1. ΔrGmø= kJ·mol-1. Kø= exp(- ΔrGmø/RT) =")
28
3.3.3 有纯态凝聚相参加的理想气体化学反应 有气相和凝聚相(液相、固体)共同参与的反应称为复相化学反应。只考虑凝聚相是纯态的情况,纯态的化学势就是它的标准态化学势,所以复相反应的热力学平衡常数只与气态物质的压力有关。 例如,有下述反应,并设气体为理想气体: 称为CaCO3 的解离压力。

29
解离压力(dissociation pressure)
某固体物质发生解离反应时,所产生气体的压力,称为解离压力,显然这压力在定温下有定值。 如果产生的气体不止一种,则所有气体压力的总和称为解离压。 例如: 解离压力 则热力学平衡常数:

30
多相反应 aA(l)+eE(g)=gG(s)+hG(g)
纯凝聚相:μA=μA Ø , μG=μGØ 而 μE=μEØ+RTln(pE/pEØ), μH=μHØ+RTln(pH/pHØ) 则 ΔrGm=- aμA- eμE+ gμG+ hμH
, μH=μHØ+RTln(pH/pHØ) 则 ΔrGm=- aμA- eμE+ gμG+ hμH.")
31
3.4 平衡常数的测定和平衡转化率的计算 平衡常数的测定 平衡转化率的计算

32
3.4.1 平衡常数的测定 (1)物理方法 直接测定与浓度或压力呈线性关系的物理量,如折光率、电导率、颜色、光的吸收、定量的色谱图谱和磁共振谱等,求出平衡的组成。这种方法不干扰体系的平衡状态。 (2)化学方法 用骤冷、抽去催化剂或冲稀等方法使反应停止,然后用化学分析的方法求出平衡的组成。
化学方法 用骤冷、抽去催化剂或冲稀等方法使反应停止,然后用化学分析的方法求出平衡的组成。")
33
3.4.2 平衡转化率的计算 平衡转化率又称为理论转化率,是达到平衡后,反应物转化为产物的百分数。
3.4.2 平衡转化率的计算 平衡转化率又称为理论转化率,是达到平衡后,反应物转化为产物的百分数。 工业生产中称的转化率是指反应结束时,反应物转化为产物的百分数,因这时反应未必达到平衡,所以实际转化率往往小于平衡转化率。

34
3.5 标准状态下反应的吉布斯能变化及化合物的标准生成吉布斯能
3.5 标准状态下反应的吉布斯能变化及化合物的标准生成吉布斯能 标准状态下反应的吉布斯能变化 化合物的标准生成吉布斯能 离子的标准摩尔生成吉布斯自由能 数值的用处

35
3.5.1 标准状态下反应的吉布斯能变化 在温度T时,当反应物和生成物都处于标准态,发生反应进度为1 mol的化学反应Gibbs自由能的变化值,称为标准摩尔反应吉布斯自由能变化(standard molar Gibbs energy change), 用△rGmØ(T)表示。 △rGmØ的用途: (1)计算热力学平衡常数
计算热力学平衡常数.")
36
(2) 计算实验不易测定的平衡常数 例如,求 的平衡常数 (1) - (2) 得(3)
 计算实验不易测定的平衡常数 例如,求 的平衡常数 (1) - (2) 得(3)")
37
3.近似估计反应的可能性 只能用(ΔrGm)T,p,Wf=0 判断反应的方向。但是,当ΔrGmØ的绝对值很大时,基本上决定了ΔrGm的值,所以可以用来近似地估计反应的可能性。 一般来说 △rGmø>42 kJ·mol-1 反应不能进行 △rGmø<-42 kJ·mol-1 反应能自发进行

38
一般有以下几种方法计算化学反应的△rGmØ
(1)热化学方法:△rGmØ=△rHmØ-T△rSmØ (2)实验测定:测定平衡常数,求△rGmØ (3)利用标准生成吉布斯能计算 (4)电化学方法:设计电池反应,△rGmØ=-nEØF
热化学方法:△rGmØ=△rHmØ-T△rSmØ. (2)实验测定:测定平衡常数,求△rGmØ. (3)利用标准生成吉布斯能计算. (4)电化学方法:设计电池反应,△rGmØ=-nEØF.")
39
3.5.2 化合物的标准生成吉布斯能 因为吉布斯自由能的绝对值不知道,所以只能用相对标准,即将标准压力下稳定单质(包括纯的理想气体,纯的固体或液体)的生成吉布斯自由能看作零,则: 在标准压力下,由稳定单质生成1 mol化合物时吉布斯自由能的变化值,称为该化合物的标准生成吉布斯自由能,用下述符号表示: △fGmØ(化合物,物态,温度) 通常在 K时的值有表可查。
 通常在 K时的值有表可查。")
40
3.5.3 离子的标准摩尔生成吉布斯自由能 有离子参加的反应,主要是电解质溶液。溶质的浓度主要用质量摩尔浓度表示,用的标准态是mØ=1mol·kg-1 且具有稀溶液性质的假想状态,这时规定的相对标准为: 由此而得到其他离子的标准摩尔生成吉布斯自由能的数值。

41
数值的用处 ΔfGmØ的值在定义时没有规定温度,通常在 K时的数值有表可查,利用这些表值,我们可以: (1) 计算任意反应在 K时的ΔfGmØ
 计算任意反应在 K时的ΔfGmØ")
42
(2)判断反应的可能性。在有机合成中,可能有若干条路线,用计算ΔfGmØ的方法,看那条路线的值最小,则可能性最大。若ΔfGmØ的值是一个很大的正数,则该反应基本上不能进行。
(3)用ΔfGmØ值求出热力学平衡常数KpØ值。根据KpØ与温度的关系,可以决定用升温还是降温的办法使反应顺利进行。
用ΔfGmØ值求出热力学平衡常数KpØ值。根据KpØ与温度的关系,可以决定用升温还是降温的办法使反应顺利进行。")
43
3.6 温度对平衡常数的影响 根据吉布斯-核姆霍兹公式,在标准状态下:

44
van’t Hoff 公式的微分式 化学反应等压方程式 对吸热反应,ΔrHmØ>0,升高温度,KpØ增加,对正反应有利。 对放热反应,ΔrHmØ>0,升高温度,KpØ降低,对正反应不利。

45
若温度区间不大,ΔrHmØ可视为常数,得定积分式为:
这公式常用来从已知一个温度下的平衡常数求出另一温度下的平衡常数。 若ΔrHmØ值与温度有关,则将关系式代入微分式进行积分,并利用表值求出积分常数。

46
例 环己烷和甲基环戊烷之间有异化作用: C6H12(l)=C5H9CH3(l) 异构化反应的平衡常数与温度有如下关系: lnK Ø = /T,试求25℃异构化反应的熵变。 lnKØ=-△rHmØ / RT + C △rGmØ=△rHmØ-T△rSmØ

48
当理想气体用浓度表示时,因为p = cRT ,可以得到
这个公式在气体反应动力学中有用处。

49
3.7 其他因素对平衡的影响 3.7.1 压力对化学平衡的影响
3.7 其他因素对平衡的影响 3.7.1 压力对化学平衡的影响 根据Le chatelier原理,增加压力,反应向体积减小的方向进行。这里可以用压力对平衡常数的影响从本质上对原理加以说明。 对于理想气体, KpØ仅是温度的函数

50
因为 所以 KcØ也仅是温度的函数。

51
对理想气体 Kx与压力有关, ,气体分子数减少,加压,反应正向进行,反之亦然。

52
对凝聚相反应 ΔνB*>0,体积增加,增加压力,KaØ下降,对正向反应不利,反之亦然。 在压力不太大时,因ΔνB*值不大,压力影响可以忽略不计。

53
3.7.2 惰性气体对化学平衡的影响 惰性气体不影响平衡常数值,当 不等于零时,加入惰性气体会影响平衡组成。
3.7.2 惰性气体对化学平衡的影响 惰性气体不影响平衡常数值,当 不等于零时,加入惰性气体会影响平衡组成。 例如: ,增加惰性气体, 值增加,括号项下降。因为 KpØ为定值,则 项应增加,产物的含量会增加。 对于分子数增加的反应,加入水气或氮气,会使反应物转化率提高,使产物的含量增加。

54
3.7.3 同时平衡 在一个反应体系中,如果同时发生几个反应,当到达平衡态时,这种情况称为同时平衡。
3.7.3 同时平衡 在一个反应体系中,如果同时发生几个反应,当到达平衡态时,这种情况称为同时平衡。 在处理同时平衡的问题时,要考虑每个物质的数量在各个反应中的变化,并在各个平衡方程式中同一物质的数量应保持一致。

55
例题:600 K时,CH3Cl(g)与H2O(g)发生反应生成 CH3OH,继而又生成(CH3)2O,同时存在两个平衡:
已知在该温度下,Kp,1Ø= ,Kp,2Ø=10.6。今以等量的CH3Cl和H2O开始,求CH3Cl的平衡转化率。

56
解:设开始时CH3Cl和H2O的摩尔分数为1.0,到达平衡时,生成HCl的摩尔分数为x,生成(CH3)2O为y,则在平衡时各物的量为:
2O为y,则在平衡时各物的量为:")
57
因为两个反应的 都等于零,所以KpØ=Kx
将两个方程联立,解得x=0.048,y=0.009 。CH3Cl 的转化率为0.048或4.8 。

58
3.8 反应的耦合 3.8.1 耦合反应(coupling reaction)
3.8 反应的耦合 3.8.1 耦合反应(coupling reaction) 设体系中发生两个化学反应,若一个反应的产物在另一个反应中是反应物之一,则这两个反应称为耦合反应。例如: 利用△rGmØ值很负的反应,将△rGmØ值负值绝对值较小甚至略大于零的反应带动起来。
 设体系中发生两个化学反应,若一个反应的产物在另一个反应中是反应物之一,则这两个反应称为耦合反应。例如: 利用△rGmØ值很负的反应,将△rGmØ值负值绝对值较小甚至略大于零的反应带动起来。")
59
3.8.2 耦合反应的用途: 例如:在 K时: 反应(1)、(2)耦合,使反应(3)得以顺利进行。
、(2)耦合,使反应(3)得以顺利进行。")
60
3.8.3 生化反应的平衡(生物能力学 bioenergetics)
a. 生化反应的标准态 规定: H+ 的标准态为 c(H+)=10-7 mol·dm-3 即 pH=7 的状态. 在生化反应中,凡涉及H+的反应,标准Gibbs函数变用ΔGmØ表示. ΔGmØ ΔGmØ
=10-7 mol·dm-3. 即 pH=7 的状态. 在生化反应中,凡涉及H+的反应,标准Gibbs函数变用ΔGmØ表示. ΔGmØ. ΔGmØ.")
61
若 T = K 注意: ν是 H+ 的化学计量数,可正可负. b.生化反应的耦合 从能量的角度来看,生物系统中的反应可分为两大类:吸能反应与放能反应。前者如肌肉收缩时的化学反应和蛋白质、核酸等的合成反应。后者如光合作用和呼吸作用。

62
OH OH OH O NH2 C N N C CH HC C N N HO - P - O - P - O - P - O - CH2
腺苷 NH2 C N N C CH HC C N N 三磷酸腺苷( ATP ) (adenosine triphosphophats) OH OH OH HO - P - O - P - O - P - O - CH2 O O O O C H H C H C C H OH OH ADP AMP ATP
 (adenosine triphosphophats) OH OH OH. HO - P - O - P - O - P - O - CH2. O O O. O. C H H C. H C C H. OH OH. ADP. AMP. ATP.")
63
ADP + HPO42- + H (1) ATP + H2O ATP: 高能代谢物 C6H12O6 + HPO42- →C6H11O6PO32- +H2O ΔGm = 13.4 kJ.mol-1 与(1)偶联 ATP + C6H12O6 →ADP + C6H11O6PO32- ΔGm = kJ.mol-1
偶联. ATP + C6H12O6 →ADP + C6H11O6PO32- ΔGm = kJ.mol-1.")
64
在细胞内谷氨酸盐与NH4+合成谷酰胺的过程:
谷氨酸盐+ NH4+ →谷酰胺 ΔGm = kJ.mol-1 ATP + →ADP + H2PO4- 谷氨酸盐+ NH4+ +ATP →谷酰胺+ADP + H2PO4- ATP的合成 PEP(磷酸烯醇丙酮酸)+ H2O = 丙酮酸 + Pi ΔGm = kJ.mol-1 ADP + Pi = ATP + H2O PEP + ADP =丙酮酸 + ATP ΔGm = kJ.mol-1
+ H2O = 丙酮酸 + Pi. ΔGm = kJ.mol-1. ADP + Pi = ATP + H2O. PEP + ADP =丙酮酸 + ATP ΔGm = kJ.mol-1.")
65
高自由能化合物 (葡萄糖) O ADP + Pi 分解代谢 生物合成 物质迁移 肌肉收缩 CO2 + H2O ATP














 3 酸碱的强弱 二、水的自偶电离平衡>")






是多羟基的醛、酮或其简单衍生物以及能水解产生上述产物的化合物的总称。>")
