Download presentation
Presentation is loading. Please wait.

1
腓骨肌萎缩症一例汇报 航空总医院 杨琼 指导医师;张洁 2014-05-09

2
目录

3
病例简介 男性, 21 岁,以 “ 四肢远端无力肌肉萎缩并进行性加重 5 年 ” 入院。 现病史: 发病过程 : 5 年前患者自觉双侧拇指无力,持物不稳,并发现双侧大 鱼际肌及骨间肌逐渐萎缩,以右手更为明显,书写时右手抖动,患者 未予重视, 3 年前开始明显感觉双足无力,不能屈曲,以右足为主, 上楼时较明显,不能穿拖鞋,偶有肌肉跳动,部位不固定,发作不频 繁。 就诊经历:先后就诊于河南某医院及乌鲁木齐军区总医院,未系统诊 断及治疗,于 2-4 就诊于我院门诊, 查体 : 冈上肌 / 冈下肌萎缩, 双手大鱼 际肌萎缩, 大拇指力弱, 双足背无力, 四肢近端肌力 5 级, 脚尖走路差, 脚跟 走路不能, 四肢腱反射偏低, 查血沉正常、类风湿因子、 ASO 及 CRP 正常, 肌酸激酶 2136 U/L , CK-MB 66.4 U/L ,为进一步诊治,门诊以 “ 神经肌 肉病 ” 收入我科。

4
病例简介 既往史: 2004 年过敏性紫癜病史,就诊于三门峡中医院治疗后 (具体不详)好转。 个人史: 今年毕业于河南漯河专科医学院口腔专业。生长发育史 无殊。 家族史: 母亲双足不能背屈,双侧大小鱼际肌萎缩,近 5 年来觉四 肢远端无力,无明显加重,无弓形足, 1 姐 25 岁无明显异常。
好转。 个人史: 今年毕业于河南漯河专科医学院口腔专业。生长发育史 无殊。 家族史: 母亲双足不能背屈,双侧大小鱼际肌萎缩,近 5 年来觉四 肢远端无力,无明显加重,无弓形足, 1 姐 25 岁无明显异常。")
5
病例简介 入院时查体: 神清语利,双侧水平旋转眼震, 上肢远端 3-4 级,大小鱼际肌、 第一骨间肌萎缩明显,蚓状肌 尚可,肱二头肌、三角肌肥大, 肱三头肌萎缩,弓形足,股四 头肌外侧头肥大,胫前肌萎缩, 双足跖屈 3-4 级,背屈 0 级,上肢 腱反射( + ),下肢腱反射未引 出。跨阈步态,不能下蹲,一 字步不行。
,下肢腱反射未引 出。跨阈步态,不能下蹲,一 字步不行。")
6
病例简介

7
辅助检查 检验结果: 血、尿、便常规基本正常。 入院时 CK 2136 U/L , CK-MB 66.4 U/L ,复查 CK 421.0U/L ; 垂体泌乳素 :23.50ng/ml ; 乳酸 :1.1mmol/L ( 0.5 ~ 1.7mmol/L ) ; 【超声心动图】二尖瓣少量返流,射血分数 59%. 【心电图】窦性心动过缓伴不齐, ST 段改变( J 点抬高), T 波改变 (高尖)。 【肌电图及诱发电位】该患者及母亲均表现为周围神经源性损害,以 轴索损害为主。 患者因经济原因拒绝头颅 MRI 检查。
, T 波改变 (高尖)。 【肌电图及诱发电位】该患者及母亲均表现为周围神经源性损害,以 轴索损害为主。 患者因经济原因拒绝头颅 MRI 检查。.")
8
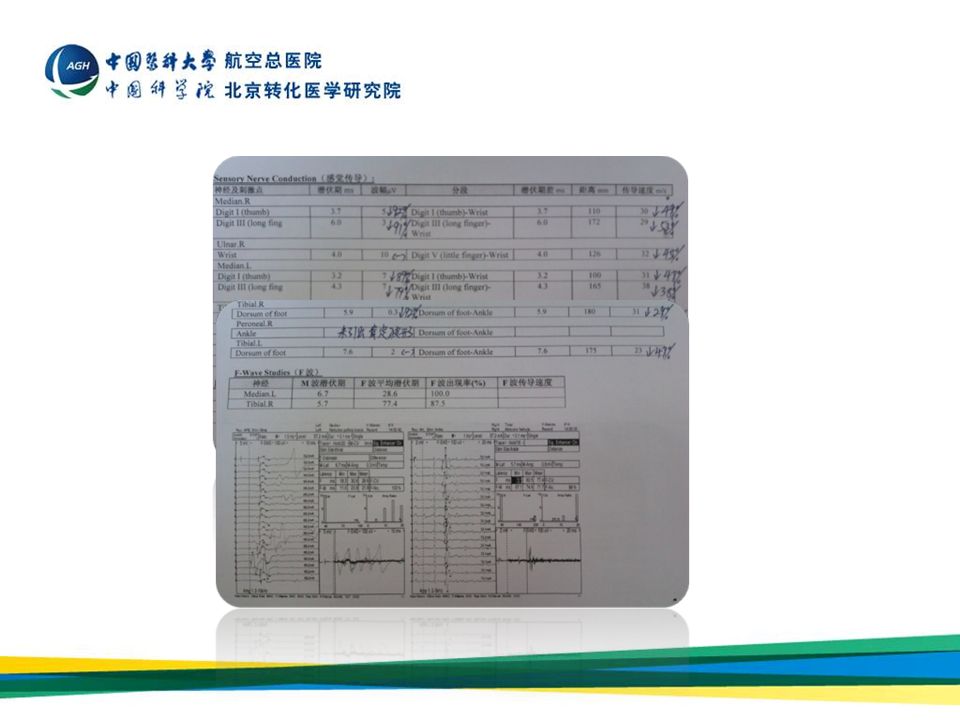
正中神经传 导速度

10
定位定性 定位诊断: 患者四肢远端无力,肌肉萎缩并进行性加重,查体 腱反射减弱,定位于下运动神经元,结合肌电图周围神经源性损害, 以轴索损害为主,故主要定位于周围神经。 定性诊断: 患者青年男性,慢性起病,主要表现为四肢远端无 力,肌肉萎缩并进行性加重,查体腱反射减弱,结合母亲类似病史, 故首先考虑遗传性神经肌肉病。且患者症状轻辅助检查重,支持遗传 性疾病。

11
鉴别诊断 遗传性周围神经病 1 、家族性淀粉样多神经病:通常 25-45 岁起病,以下肢感觉障碍和自 主神经功能障碍为早期特征,需借助神经活检及 DNA 鉴别; 2 、遗传性共济失调伴肌萎缩:又称 Roussy-levy 综合征,临床症状类似 于腓骨肌萎缩症,但尚有站立不稳、步态蹒跚、手震颤等; 3 、遗传性压迫易感性神经病( HPNN ):因有肌无力、萎缩和传导速 度减慢需与 CMT 鉴别,但常发生于轻微牵拉、压迫或外伤后反复出现 肌无力、萎缩。麻木、踝反射消失、弥漫性神经传导速度减慢,神经 火箭为节段性脱髓鞘和腊肠样结构改变,预后良好。
:因有肌无力、萎缩和传导速 度减慢需与 CMT 鉴别,但常发生于轻微牵拉、压迫或外伤后反复出现 肌无力、萎缩。麻木、踝反射消失、弥漫性神经传导速度减慢,神经 火箭为节段性脱髓鞘和腊肠样结构改变,预后良好。")
12
鉴别诊断 4 、遗传性共济失调多发神经炎样病( Refsum 病 ) 对称性肢体无力和肌 萎缩及腱反射减弱需与 CMT 鉴别,但本病除有多发性周围神经损害外, 还有小脑性共济失调、夜盲、视网膜色素变性及 CSF 脑脊液增高; 5 、慢性进行性远端型脊肌萎缩症:该病的肌萎缩及病程类似 CMT , 但伴有肌肉跳动、 EMG 示前角损害,无感觉传导障碍可与 CMT 鉴别; 6 、肯尼迪病: X 染色体连锁的遗传性运动神经元病,主要见于中年男 性,表现为缓慢进展的延髓损害和肢体肌肉无力、萎缩和束颤,可有 构音不清和吞咽困难,无上运动神经元损害、缓慢的病程及近端对称 形式的肌无力有助于鉴别诊断,肯尼迪病还有雄激素不足的表现,包 括男性乳房女性化。睾丸萎缩和阳痿。确诊需基因检测。
 对称性肢体无力和肌 萎缩及腱反射减弱需与 CMT 鉴别,但本病除有多发性周围神经损害外, 还有小脑性共济失调、夜盲、视网膜色素变性及 CSF 脑脊液增高; 5 、慢性进行性远端型脊肌萎缩症:该病的肌萎缩及病程类似 CMT , 但伴有肌肉跳动、 EMG 示前角损害,无感觉传导障碍可与 CMT 鉴别; 6 、肯尼迪病: X 染色体连锁的遗传性运动神经元病,主要见于中年男 性,表现为缓慢进展的延髓损害和肢体肌肉无力、萎缩和束颤,可有 构音不清和吞咽困难,无上运动神经元损害、缓慢的病程及近端对称 形式的肌无力有助于鉴别诊断,肯尼迪病还有雄激素不足的表现,包 括男性乳房女性化。睾丸萎缩和阳痿。确诊需基因检测。")
13
鉴别诊断 遗传性肌肉病: 远端型肌营养不良:四肢远端无力、肌萎缩、渐向上发展与 CMT 鉴别, 但成年起病,肌电图示肌源性损害,运动传导速度正常可鉴别。患者 入院时 CK 较高,需与此病鉴别,但复查后 CK 较前有所下降,且肌电图 示神经源性损害较重,故不考虑。 平山病为良性自限性运动神经元疾病,青春早期隐袭起病的手及前臂 远端肌肉无力,随病变进展逐渐出现相应肌群萎缩,多为单侧损害, 部分也可表现为不对称双侧损害。

14
基因及病理

15
目前国内 GJB1 家系研究 Asn2Lys 天冬酰 胺 — 赖氨酸 Glu208Lys 谷氨 酸 — 赖氨酸 Ile127Phe 异亮氨 酸 — 苯丙氨酸 Arg15Glu 精氨 酸 — 谷氨酰胺

16
目录

17
腓骨肌萎缩症 (Charcot-Marie-Tooth disease,CMT) 最常见的具有高度临床和遗传异质性的周围神经单基因遗传病 患病率 1/2500 疾病历史: Charcot , Marie 和 Tooth 于 1886 年首先报道。 临床症状: 儿童和青少年起病; 进行性对称性肢体远端肌无力和肌萎缩; 感觉障碍和腱反射减退或消失; 高弓足,脊柱侧弯和骨骼畸形。
 最常见的具有高度临床和遗传异质性的周围神经单基因遗传病 患病率 1/2500 疾病历史: Charcot , Marie 和 Tooth 于 1886 年首先报道。 临床症状: 儿童和青少年起病; 进行性对称性肢体远端肌无力和肌萎缩; 感觉障碍和腱反射减退或消失; 高弓足,脊柱侧弯和骨骼畸形。")
18
CMT 电生理病理分型 脱髓鞘型( CMT1 ) 轴索型 (CMT2) 中间型 正中神经运动传导速度低于 38m / s 显著的髓鞘异常 ( 节段性脱髓鞘,雪 旺细胞增生,呈 “ 洋葱头 ” 样改变 正中神经运动传导速度大于 38m / s 慢性轴索变性和再生 ( 轴索变性和有 髓纤维减少,神经再生簇形成 ) 正中神经传导速度介于 25 ~ 45m / s , 脱髓鞘和轴索变性
 轴索型 (CMT2) 中间型 正中神经运动传导速度低于 38m / s 显著的髓鞘异常 ( 节段性脱髓鞘,雪 旺细胞增生,呈 洋葱头 样改变 正中神经运动传导速度大于 38m / s 慢性轴索变性和再生 ( 轴索变性和有 髓纤维减少,神经再生簇形成 ) 正中神经传导速度介于 25 ~ 45m / s , 脱髓鞘和轴索变性")
19
CMT 基因分型 J. Berciano,et al.Neurología. 2012 37 loci with 31 cloned genes have been iden-tified using genetic linkage analysis

20
CMT 基因分型特殊临床表现 CMT1D 脑神经受损 CMT1X 中枢神经受损 CMT2A 视神经萎缩 CMT2C 声带麻痹及呼吸受累 CMT2D 上肢受累严重 CMT4B2 早发性青光眼 CMT4C 重度脊柱侧弯 CMT4F 感觉缺失明显 HMNSR 听力丧失

21
CMT 诊断流程 J. Berciano,et al.Neurología. 2012

22
CMT 发病机制 a)alteration of the development and maintenance of myelin; b)alteration of the biosynthesis and degradation of proteins; c) alteration of the endocytosis and dynamics of membranes; d)alteration of the axonal cytoskeleton; e) seipinopathies ; f) channelopathies by mutation of TRPV4.
alteration of the development and maintenance of myelin; b)alteration of the biosynthesis and degradation of proteins; c) alteration of the endocytosis and dynamics of membranes; d)alteration of the axonal cytoskeleton; e) seipinopathies ; f) channelopathies by mutation of TRPV4.")
23
目录

24
CMT1X 的诊断 发病率:第 2 位, 占所有 CMT 患者的 7% ~ 11%, 仅次于 CMT1A 型。 遗传学特征: X- 连锁的显性遗传模式, 无男传男现象, 且半合子男性患者较杂合子女 性患者发病年龄较早, 临床表现较重。男性多在 5 ~ 20 岁之间发病, 而 女性多在 20 岁之后发病 病理学特征(腓肠肌活检): 脱髓鞘的改变 + 轴索丢失(年龄相关的有髓纤维的丢失以及再生轴索 簇数目的增加); 薄髓鞘常见(慢性脱髓鞘和髓鞘再生或者轴索再生后的髓鞘再生); 洋葱球样结构不常见( Ile127Ser 突变的病例中, 洋葱球形成是其主要 的病理特征)
: 脱髓鞘的改变 + 轴索丢失(年龄相关的有髓纤维的丢失以及再生轴索 簇数目的增加); 薄髓鞘常见(慢性脱髓鞘和髓鞘再生或者轴索再生后的髓鞘再生); 洋葱球样结构不常见( Ile127Ser 突变的病例中, 洋葱球形成是其主要 的病理特征)")
25
CMT1X 的电生理(中间型)
")
26
CMT1X 的特殊临床学表现 累及中枢神经系统 临床表现:急性播散性脑脊髓炎 (ADEM) 样发作, 听力丧失, 精神育迟滞, 扫视性眼球运动, 暂时性下肢轻瘫或单瘫, 椎体束征阳性, 共济失调, 咽 反射消失, 发音困难, 吞咽困难, 亚急性呼吸窘迫综合症等。 头颅 MRI :脑白质损害, 可出现在半卵圆中心部、胼胝体压部、内囊、 小脑中脚、顶枕部等, 表现为 T2 加权信号增强及脱髓鞘改变。发作期 过后, MRI 可部分或完全恢复正常。还有患者出现 MRI 异常而无相应临 床表现。
 样发作, 听力丧失, 精神育迟滞, 扫视性眼球运动, 暂时性下肢轻瘫或单瘫, 椎体束征阳性, 共济失调, 咽 反射消失, 发音困难, 吞咽困难, 亚急性呼吸窘迫综合症等。 头颅 MRI :脑白质损害, 可出现在半卵圆中心部、胼胝体压部、内囊、 小脑中脚、顶枕部等, 表现为 T2 加权信号增强及脱髓鞘改变。发作期 过后, MRI 可部分或完全恢复正常。还有患者出现 MRI 异常而无相应临 床表现。")
27
CMT1X 累及中枢神经系统影像学变化 Robert A. Taylor,et al. 2003 neurologyG H :eleven weeks follow up

28
GJB1 基因致病机制 GJB1 基因:位于 X 染色体的 Xq13.1 位点, 编码缝隙连接蛋白 CX32 。 Connexin32(CX32) : 4 个跨膜域, 2 个胞外环, 1 个胞内环, 一个 C 端和一个 N 端均位于胞内,形 成缝隙连接,对施万细胞的信号传导其重要作用。 广泛表达于肝脏, 胰腺外分泌部, 中枢神经系统的少突胶质细胞、星形 胶质细胞及神经元, 周围神经的施万细胞以及胃肠道的上皮组织。 髓鞘: Ranvier node 和 Schmidt-Lanterman 切迹。
 : 4 个跨膜域, 2 个胞外环, 1 个胞内环, 一个 C 端和一个 N 端均位于胞内,形 成缝隙连接,对施万细胞的信号传导其重要作用。 广泛表达于肝脏, 胰腺外分泌部, 中枢神经系统的少突胶质细胞、星形 胶质细胞及神经元, 周围神经的施万细胞以及胃肠道的上皮组织。 髓鞘: Ranvier node 和 Schmidt-Lanterman 切迹。")
29
CX32 的分子结构 four transmembrane domains, one intracellular and two extracellular loops, an amino- and a carboxy- terminal cytoplasmic tail. The GJB1 mutations of the coding region are indicated, more than 400 altogether since 1993 ( <1000 Da). Kleopas A. Kleopa,et al. brain research 2013
. Kleopas A. Kleopa,et al. brain research")
30
CX32 形成六聚体结构 髓鞘的增殖、 分化、凋亡 连接细胞骨架 和基底膜,增 强髓鞘稳定性 PMP22/MPZ 复 合体,髓磷脂 合成 转录调控因子, 调节 PMP22 MPZ CX32 合成

31
CX32 致病机理 缝隙连接 :不能形成缝隙连接或形成有缺陷的缝隙连接 基因功能缺失( loss of function) a) 不同的基因突变所引起临床表现的严重程度相似, 尤其是基因整个编码 区的缺失所引起的症状与其他类型突变相似 b) 在 CX32 基因敲除小鼠中能够看到同 CMT1X 临床表现相似的进行性周围 感觉运动神经病 显性负性作用 (dominant-negative effect) a) 中枢神经系统: CX32 和 CX47 单基因敲除均无中枢受累。 CX32 和 CX47 联合敲除的小鼠表现有中枢神经的受累, CX32 和 CX47 两种缝隙连接蛋 白缺乏的小鼠在出生后 6 周死于显著的中枢髓鞘的异常 b) 中枢神经系统: Cx32 or Cx26 (R142W and R75W) c) 周围神经系统: CX32 和 CX29
 a) 不同的基因突变所引起临床表现的严重程度相似, 尤其是基因整个编码 区的缺失所引起的症状与其他类型突变相似 b) 在 CX32 基因敲除小鼠中能够看到同 CMT1X 临床表现相似的进行性周围 感觉运动神经病 显性负性作用 (dominant-negative effect) a) 中枢神经系统: CX32 和 CX47 单基因敲除均无中枢受累。 CX32 和 CX47 联合敲除的小鼠表现有中枢神经的受累, CX32 和 CX47 两种缝隙连接蛋 白缺乏的小鼠在出生后 6 周死于显著的中枢髓鞘的异常 b) 中枢神经系统: Cx32 or Cx26 (R142W and R75W) c) 周围神经系统: CX32 和 CX29")
32
GJB1 中 R142W 的突变导致 CX32 异常表达 When transfected into otherwise connexin-deficient PC12 cells, R142W, showed no detectable staining on the plasma membrane but instead appeared to be defective in intracellular transport (De-schenes et al., 1997). The R142W Cx32 mutant also accumulated intracellularly when expressed under the control of the myelin-specific Po promotor in the Schwann cells of either Cx32 knock-out or otherwise normal mice (Bone et al., 1997). R142W transgenic mice showed retention of the mutant protein in the Golgi and developed a mild demyelinating neuropathy (Jeng et al., 2006 ). the presence of the mutant Cx32 reduced the level of the endogenous mouse Cx32, indicating that R142W has a dominant-negative effect on wild-type Cx32. R142W, have developed the striking picture of an acute, transient encephalopathy associated with MRI changes in CNS myelin.
. R142W transgenic mice showed retention of the mutant protein in the Golgi and developed a mild demyelinating neuropathy (Jeng et al., 2006 ). the presence of the mutant Cx32 reduced the level of the endogenous mouse Cx32, indicating that R142W has a dominant-negative effect on wild-type Cx32. R142W, have developed the striking picture of an acute, transient encephalopathy associated with MRI changes in CNS myelin..")
33
目录

34
CMT 治疗策略 Vitamins and Fatty Acids Progesterone Antagonists ( A more severe neuropathy developed in transgenic CMT1A rats treated with daily subcutaneous injections of progesterone (20 mg/kg), with increased levels of PMP22 and MPZ mRNA levels in peripheral nerves) Curcumin (increases in the number and size of myelinated axons). Molecular Genetic and Cellular Therapies

35
病例总结 周围神经损害 轴索为主 R142W 突变导 致 GJB1 编码 CX32 表达异常, 导致脱髓鞘和 轴索损害

Similar presentations


. 免疫缺陷病 (Immunodeficiency diseade,IDD) : 由免疫系统中任何一个成分在发生、发 育和成熟过程中的缺失或功能不全而导致免 疫功能障碍所引起的疾病。 免疫缺陷病分为 : 先天性 />")

染色体结构不同; ( 2 )原核生物具有正调控和负调控并重的特点,真核 生物目前已知的主要是正调控; ( 3 )原核生物的转录和翻译是相偶联的,真核生物的.>")
 损害的特征。感觉系统一般不受侵犯。>")
>")
 运动神经元病 上+下运动神经元受累 上运动神经元受累 下运动神经元受累 单肢肌萎缩 (Hirayama 病 ) 副肿瘤运动神经元病 Hopkins 综合症 后脊髓灰质炎综合症 多灶运动神经病 急性轴索运动神经病 卟啉病 中毒.>")


>")



>")
 全世界最杰出的物理学家之一史蒂芬·霍金>")




