Download presentation
Presentation is loading. Please wait.

1
第五章 非线性药物动力学 中国药科大学研究生讲义 主讲:陈西敬 教授

2
非线性药动学的定义 临床上某些药物存在非线性的吸收或分布(如抗坏血酸,甲氧萘丙酸);还有一些药物以非线性的方式从体内消除,过去发现有水杨酸、苯妥英钠和乙醇等。这主要是由于酶促转化时药物代谢酶具有可饱和性,其次肾小管主动转运时所需的载体也具有可饱和性,所以药物在体内的转运和消除速率常数呈现为剂量或浓度依赖性(dose dependent),此时药物的消除呈现非一级过程,一些药动学参数如药物半衰期、清除率等不再为常数,AUC、Cmax等也不再与剂量成正比变化。上述这些情况在药动学上被称之为非线性动力学(nonlinear pharmacokinetics)
;还有一些药物以非线性的方式从体内消除,过去发现有水杨酸、苯妥英钠和乙醇等。这主要是由于酶促转化时药物代谢酶具有可饱和性,其次肾小管主动转运时所需的载体也具有可饱和性,所以药物在体内的转运和消除速率常数呈现为剂量或浓度依赖性(dose dependent),此时药物的消除呈现非一级过程,一些药动学参数如药物半衰期、清除率等不再为常数,AUC、Cmax等也不再与剂量成正比变化。上述这些情况在药动学上被称之为非线性动力学(nonlinear pharmacokinetics)")
3
研究目的与意义 非线性药物动力学的研究对临床上一些治疗指数较窄的药物(如苯妥英等)来说意义非常重大,了解它们的药动学特征,有利于避免出现药物不良反应和保证临床疗效。目前新药的药动学研究中规定,必须对药动学性质的进行研究,即研究不同剂量下药物的药动学行为是否发生变化,有时还需研究药物在中毒剂量下的药动学性质。
来说意义非常重大,了解它们的药动学特征,有利于避免出现药物不良反应和保证临床疗效。目前新药的药动学研究中规定,必须对药动学性质的进行研究,即研究不同剂量下药物的药动学行为是否发生变化,有时还需研究药物在中毒剂量下的药动学性质。")
4
第一节 非线性药物消除的动力学特征 一、非线性药物动力学的表达方法
药物的生物转化,肾小管主动分泌及胆汁的分泌,通常需要酶或载体系统参与,这些系统相对于底物有较高的专属性,且能力有一定的限度,即具有饱和性,通常这些饱和过程的动力学可以用米氏方程来表征 C指血药浓度;Vm为酶促反应的最大速率,其单位为浓度/时间;Km为米氏速率常数,其单位为浓度,其定义为酶促反应达到最大速率一半时的血药浓度。

5
浓度与消除速度的关系 1.当剂量或浓度较低时,C《Km, 此时米氏方程 分母中的C可以忽略不计,则上式可简化为 dC/dt = k´C
此时相当于一级过程。由图7-1可见,低浓度时logC-t为一直线。 2.当剂量或浓度较高时,C》Km 分母中的Km可以忽略不计,则米氏方程可简化为: dC/dt=Vm 此时相当于零级过程,由图7-1可见,高浓度时logC几乎不随t变化,原因是酶的作用出现饱和,此时t1/2与初浓度成正比关系 t1/2=C0/(2Vm) 即t1/2随C0而递增。 3.当剂量或浓度适中时,则米氏方程形式不变此时药物在体内的消除呈现为混合型。
 即t1/2随C0而递增。 3.当剂量或浓度适中时,则米氏方程形式不变此时药物在体内的消除呈现为混合型。")
6
二、动力学特征 据上所述,可将非线性药物动力学的动力学特征总结如下: 1.高浓度时为零级过程。 2.低浓度时为近似的一级过程。
3.消除速率和半衰期不再为常数,而与初 浓度C0有关。 4.AUC与剂量不成比例。

7
三、非线性药物动力学的鉴别方法 1.LogC—t图形观察法

8
2.面积法 对同一受试者给予不同的剂量,分别计算AUC值,若AUC与X0间呈比例,说明为线性,否则为非线性。若AUC随剂量增加较快,可考虑为非线性消除;若AUC随剂量增加较慢,血管外给药的情况下可考虑为吸收出现饱和,即非线性吸收。如图7-3所示。 图7-3 线性与非线性动力学的AUC与剂量X0间的关系

9
四、t1/2和AUC与C0间的关系 对于单室系统,按单纯饱和过程消除的药物静注后,血药浓度的经时过程可通过米氏方程的积分形式来表征: 可化为
当t=t0时, Vmt0+ i=–KmlnC0–C 上述两式相减得: Vm(t–t0)=(C0–C)+Kmln(C0/C)
=(C0–C)+Kmln(C0/C)")
10
1.半衰期t1/2 C=C0/2时,t–t0=t1/2,代入公式7-8可得: t1/2表现为浓度C0依赖性

11
2.血药浓度曲线下面积(AUC) 将米氏方程形式改变后可得: 根据t=0时,C=C0;t=∞时,C=0,对公式作定积分得:
当浓度很低时(Km 》C0/2),上式简化为
,上式简化为.")
12
此时AUC与初浓度或剂量的平方成正比,其关系为抛物线形式。
接上页 此时AUC与初浓度或剂量成正比。 当浓度很高时(Km《C0/2),上式简化为 此时AUC与初浓度或剂量的平方成正比,其关系为抛物线形式。
,上式简化为. 此时AUC与初浓度或剂量的平方成正比,其关系为抛物线形式。")
13
第二节 米氏参数的估算方法 在计算机普及应用之前,对非线性方程多采用数学变换使其直线化的方法进行解析,由于该法比较简便和直观,现在仍在使用。下述方法1-4均是根据这一原理提出的,方法5为计算机拟合法。

14
1.对米氏方程两端取倒数 以dC/dt的倒数对C的倒数作图,可得一条直线,从截距–1/Vm,斜率–Km/Vm可求得Vm和Km。 -1/Vm

15
双倒数法应用举例

16
2.对米氏方程两端取倒数后×C 以C/dC/dt对C作图,可得一条直线,斜率–1/Vm,截距为–Km/Vm
(C/dC)/dt也可化为1/d(linC)/dt的形式。
/dt也可化为1/d(linC)/dt的形式。")
17
3.采用米氏方程的等价形式 Km -Vm 以dC/dt对dC/dt/C作图可得一条斜率为Km,截距为-Vm的直线。

18
4.生物半衰期法 由半衰期公式 在静注给药后,对不同的浓度求出不同的t1/2,则由t1/2对C0作图可得斜率为1/(2Vm),截距为0.693Km/Vm的直线,由斜率和截距即可求出Km和Vm。
,截距为0.693Km/Vm的直线,由斜率和截距即可求出Km和Vm。")
19
5.计算机拟合法 以上都是变换坐标系统示离米氏方程参数的方法,也可以采用计算机以非线性最小二乘法对试验数据进行拟合求出Km和Vm。

20
第三节 非线性消除与个体化给药 个体病人的苯妥英钠浓度存在差异,其原因主要为该药在体内的代谢过程为非线性动力学性质,此外为生物利用度,药物相互作用和未遵照医嘱服药。因而苯妥英钠浓度往往需要进行监测。根据用药后消除动力学方程。 R为每天给药速率,单位为mg; Vd为分布溶积,Vm和Km的定义按前述。 多次给药后,体内药量不断增加,达稳态时,摄入和消除到达平衡,血药浓度为稳态水平Css。于是dC/dt=0,则:

21
上式经变换后可得到不同形式的方程 稳态浓度给药速率函数 该式表示,当给药速率R很小时,CSS和给药速率呈线性。但随着R增加逐步呈非线性。给药速率R接近Rmax,即酶接近饱和时,CSS急骤上升 将R对R/CSS作图得斜率为-Km截距为Rmax的直线,标出个体化病人的参数Km和Rmax值。

22
举例 某一癫痫病人,按苯妥英钠90 mg/日,经数天后测得稳态水平为3.70 μg/ml,然后改用剂量为270 mg/日,测得水平为47 μg/ml。将两次结果估标出Rmax和Km,看该病人期望的浓度CSS为15 μg/ml,试问给药速率R多大为合适。 解:

23
(1)两次结果分别代入(6. 19)式,或按R/CSS对R两点作图法,见图(6. 3),算得Km和Rmax分别为9
(1)两次结果分别代入(6.19)式,或按R/CSS对R两点作图法,见图(6.3),算得Km和Rmax分别为9.7mg/升和326 mg/日。 (2)将Km和Rmax值代入,算出剂量。
两次结果分别代入(6.19)式,或按R/CSS对R两点作图法,见图(6.3),算得Km和Rmax分别为9.7mg/升和326 mg/日。 (2)将Km和Rmax值代入,算出剂量。")
24
3.双倒数公式的应用 可将公式 转化为如下的形式:
1/CSS和1/R作图将得到斜率为Rmax/Km截为-1/Km直线。于是从群体的平均值,Rmax=10.3 mg/kg/日;Km=11.5 mg/升,可设计列线图;图(6.4)为按(6.20)式倒数刻度相对距离绘制的列线图。
为按(6.20)式倒数刻度相对距离绘制的列线图。")
25
举例: 某病人体重70 kg,给苯妥英钠100 mg,测得稳态浓度为8.2 mg/升,试按列线图,算出期望浓度CSS为15 mg/升所需的剂量。 解: 每日总剂量为100 mg×3/70 kg,即4.3 mg/kg/日,图6-4中横轴找出4.3 mg/kg/日,其纵轴交于列线圈中参考标准线上8.2处,于是沿该直线而上至纵轴的15 mg/升外,向横轴作垂线,其垂足处得需剂量5.8 mg/kg/日,或297 mg/70kg/日。

26
该方法不足之处: 因Rmax=10.3 mg/日和Km=11.5 mg/升,来自一批数据的平均值,如按上述调整后,两次浓度CSS,按上述(2)法算出Km和Rmax后再调正。然而这一方法本身一开始能有依据作初始尝试要比(2)法有其优点。
法算出Km和Rmax后再调正。然而这一方法本身一开始能有依据作初始尝试要比(2)法有其优点。")
27
第四节 非线性药物吸收 药物的吸收过程至少有两三种机制参与,被动扩散和主动转运。而主动转 运又分为主动吸收和主动外排。大多数药物的吸收是通过在小肠粘膜的被动扩散进入到血液的;一部分药物的吸收是靠主动转运完成的,如β内酰胺类抗生素和ACE抑制剂依那普利等可通过肠粘膜上皮细胞腔面的寡肽转运蛋白PEPT1的摄取而得到吸收;另一些药物在进入肠粘膜上皮细胞或血液后又通过P-gp或MRP等转运蛋白分泌至肠腔。理论上讲,无论上述哪种机制均可产生饱和情况,从而造成药物吸收的非线性现象。主动吸收或被动吸收的饱和会导致剂量增加时Cmax或AUC不按比例增加,而分泌饱和时由于被外排的药物比例减少,AUC/Dose则会随剂量的增加而增大,从而使药物的生物利用度有所提高。

28
1、抗癫痫药加巴喷丁非线性吸收的研究 加巴喷丁(Gabapentin)为一种抗癫痫药,它在体内不被代谢而以肾排泄的方式被消除,该药物口服给药后存在非线性吸收现象。Gidal等对该药的非线性吸收性质和给药间隔对吸收量的影响进行了研究。结果发现该药的吸收与剂量间存在明显的非线性关系。随着给药剂量的增加,药量被吸收的比例逐渐减少,见图所示
为一种抗癫痫药,它在体内不被代谢而以肾排泄的方式被消除,该药物口服给药后存在非线性吸收现象。Gidal等对该药的非线性吸收性质和给药间隔对吸收量的影响进行了研究。结果发现该药的吸收与剂量间存在明显的非线性关系。随着给药剂量的增加,药量被吸收的比例逐渐减少,见图所示.")
29
剂量对吸收程度的影响 为了验证增加给药次数可以提高药物的吸收量,作者对不同剂量下间隔8hr和间隔6hr给药的方法进行了比较,结果在高剂量时(4800mg/天)增加给药次数对药物的峰浓度、平均浓度、排泄量Ae和生物利用度的增加十分明显,结果见表11-1所示。作者认为通过增加给药次数可以提高该药物的生物利用度,但应同时考虑病人用药的方便性和安全性
增加给药次数对药物的峰浓度、平均浓度、排泄量Ae和生物利用度的增加十分明显,结果见表11-1所示。作者认为通过增加给药次数可以提高该药物的生物利用度,但应同时考虑病人用药的方便性和安全性.")
30
2、对头孢呋辛酯非线性吸收和非线性消除的研究
头孢呋辛酯(cefuroxime)为一种β内酰胺抗生素,静脉给药后呈二房室模型分布,主要消除途径为肾排泄并且呈现肾小管重吸收饱和现象;口服给药后该药在消化道被部分降解,吸收过程中有寡肽转运蛋白PEPT1参与,高剂量下吸收出现饱和。Carretero 等采用静脉注射和灌胃两种途径给药,以HPLC法测定给药后头孢呋辛酯的血药浓度,对该药的药动学性质进行了研究[2]。 结果在静注1.78,8.9和17.8mg 头孢呋辛钠后药物的消除动力学呈明显的非线性特征,表现为随着剂量的增加血浆清除率增大,药物的消除半衰期和平均驻留时间缩短,血药浓度曲线下面积和给药剂量的比值AUC0-∞/D由30.78 h/L减小至 h/L(见表11-2所示)。作者认为药物经肾小球滤过后在肾小管的重吸收过程中出现了饱和现象。灌服其前体药物头孢呋辛酯后药物的消除速度呈现出与静脉给药相似的剂量依赖性,而最大血药浓度与剂量间的比值Cmax/D却明显减小,同时血药浓度曲线下面积和给药剂量的比值AUC0-∞/D由7.359 h/L减小至 h/L(见表11-3所示),说明药物的吸收过程出现了饱和。
为一种β内酰胺抗生素,静脉给药后呈二房室模型分布,主要消除途径为肾排泄并且呈现肾小管重吸收饱和现象;口服给药后该药在消化道被部分降解,吸收过程中有寡肽转运蛋白PEPT1参与,高剂量下吸收出现饱和。Carretero 等采用静脉注射和灌胃两种途径给药,以HPLC法测定给药后头孢呋辛酯的血药浓度,对该药的药动学性质进行了研究[2]。 结果在静注1.78,8.9和17.8mg 头孢呋辛钠后药物的消除动力学呈明显的非线性特征,表现为随着剂量的增加血浆清除率增大,药物的消除半衰期和平均驻留时间缩短,血药浓度曲线下面积和给药剂量的比值AUC0-∞/D由30.78 h/L减小至 h/L(见表11-2所示)。作者认为药物经肾小球滤过后在肾小管的重吸收过程中出现了饱和现象。灌服其前体药物头孢呋辛酯后药物的消除速度呈现出与静脉给药相似的剂量依赖性,而最大血药浓度与剂量间的比值Cmax/D却明显减小,同时血药浓度曲线下面积和给药剂量的比值AUC0-∞/D由7.359 h/L减小至 h/L(见表11-3所示),说明药物的吸收过程出现了饱和。")
31
剂量对药动学参数的影响

32
第四节 非线性药动学的研究进展 一、最近新发现的一些非线性消除的药物
第四节 非线性药动学的研究进展 一、最近新发现的一些非线性消除的药物 近年来又有一些非线性药物代谢的新的研究报道,如抗胃酸药奥美拉唑在美洲驼羊身上试验时,静注0.2 mg/kg, 0.4 mg/kg 和 0.8 mg/kg 三种剂量的半衰期分别为0.61,0.72 和1.07 h,其AUC增大的比例超过剂量增加的比例,MRT也随剂量增加而延长。 类似的情况也在抗微生物药voriconazole、抗老年痴呆症药rivastigmine、降血脂药氟伐他汀(fluvastatin)、抗癌药表皮生长因子抗体C225[5] 、紫杉醇、DNA拓扑异构酶抑制剂NB-506等,和HIV-1逆转录酶药Efavirenz[8]等。
、抗癌药表皮生长因子抗体C225[5] 、紫杉醇、DNA拓扑异构酶抑制剂NB-506等,和HIV-1逆转录酶药Efavirenz[8]等。")
33
伏立康唑的非线性代谢 抗微生物药伏立康唑(voriconazole)主要在肝脏通过CYP2C19进行代谢[4],其代谢有弱代谢和强代谢,两种代谢者所占比例因不同人种而异。为比较口服和静注两种给药方式的安全性、耐受性和药物代谢动力学。 urkins L等研究如下:第一组受试者第一天按6mg/kg静注伏立康唑两次;之后的2-7天每日静注两次,每次3mg/kg;第二周每日口服200mg两次;另一组受试者,第一天按6mg/kg静注给药两次,接下来每日按5mg/kg静注两次,第二周每日口服两次400mg;另7名受试者第一天按6mg/kg静注两次,2-7天按4mg/kg每日静注两次,8-14天每日口服两次300mg;还有7名受试者给与对应得安慰剂。结果显示,口服伏立康唑 h后达峰浓度。伏立康唑的代谢具有可饱和性,体内代谢呈非线性药动学特征。无论静注还是口服的峰浓度(Cmax)和药时曲线下面积(AUC)都与剂量的增加程度不成比例,给药剂量增加时,药时曲线下面及(AUC)急剧增加。静注剂量增加1.7倍,Cmax和AUC分别增加2.4和3.1倍。类似的,口服剂量增加2倍,Cmax和AUC分别增加2.8和3.9倍。口服4天后达到稳态,受试者对伏立康唑耐受性良好,唾液呈现与血浆相似的非线性药动学特征。
主要在肝脏通过CYP2C19进行代谢[4],其代谢有弱代谢和强代谢,两种代谢者所占比例因不同人种而异。为比较口服和静注两种给药方式的安全性、耐受性和药物代谢动力学。 urkins L等研究如下:第一组受试者第一天按6mg/kg静注伏立康唑两次;之后的2-7天每日静注两次,每次3mg/kg;第二周每日口服200mg两次;另一组受试者,第一天按6mg/kg静注给药两次,接下来每日按5mg/kg静注两次,第二周每日口服两次400mg;另7名受试者第一天按6mg/kg静注两次,2-7天按4mg/kg每日静注两次,8-14天每日口服两次300mg;还有7名受试者给与对应得安慰剂。结果显示,口服伏立康唑 h后达峰浓度。伏立康唑的代谢具有可饱和性,体内代谢呈非线性药动学特征。无论静注还是口服的峰浓度(Cmax)和药时曲线下面积(AUC)都与剂量的增加程度不成比例,给药剂量增加时,药时曲线下面及(AUC)急剧增加。静注剂量增加1.7倍,Cmax和AUC分别增加2.4和3.1倍。类似的,口服剂量增加2倍,Cmax和AUC分别增加2.8和3.9倍。口服4天后达到稳态,受试者对伏立康唑耐受性良好,唾液呈现与血浆相似的非线性药动学特征。")
34
不同剂量下伏立康唑的药动学参数

35
二、其它因素引起的药物非线性消除现象 在作用机制研究上也注意到了药物本身以外的因素,如Ellis等发现紫杉醇的非线性消除现象与制剂中的一种赋型剂cremophor有关,该赋型剂对许多细胞毒药物的代谢具有抑制作用,在对大鼠的试验中使用了80μg和800μg两种剂量的cremophor, 发现与80μg相比,800μg的cremophor使紫杉醇的AUC增加9倍,Cl降低9倍,半衰期延长了5倍,胆汁中代谢产物的清除量由原来相当于母药量的85%降低为45%。Tagawa等对一种新型非甾体抗炎药TAK-603的体内外试验表明,引起非线性消除的原因在于其去甲基化代谢物M-I,该产物是TAK-603在大鼠与人体的主要代谢产物,对肝药酶有很强的抑制作用,可完全抑制硝本地平的氧化型代谢,所以作者推断对其母药TAK-603也会有抑制作用。

36
三、新技术在非线性药物动力学研究中的应用
在研究手段上也不断有新方法出现,如Yeh等采用稳定同位素标记和LC/MS/MS技术对抗HIV药英地那韦(Indinavir)的非线性代谢行为进行了研究。三组受试者在静注16mg 6原子氘标记药物(D6)的同时,分别静注400mg、800mg、和滴注16mg的非标记药物(D0),结果发现滴注D0 16mg对D6药动学几无影响,而静注400mg, 800mg后D6的血药浓度显著增高,且以800mg剂量为最高。作者认为该法在药物非线性代谢的研究中有很高的使用价值。
的非线性代谢行为进行了研究。三组受试者在静注16mg 6原子氘标记药物(D6)的同时,分别静注400mg、800mg、和滴注16mg的非标记药物(D0),结果发现滴注D0 16mg对D6药动学几无影响,而静注400mg, 800mg后D6的血药浓度显著增高,且以800mg剂量为最高。作者认为该法在药物非线性代谢的研究中有很高的使用价值。")
37
四、药物的非线性结合研究 除了对吸收和代谢方面的研究外,最近也注意到了蛋白结合引起的非线性代谢。如对一种新的青霉素类药物MK-826在大鼠和猴身上实验时,将剂量从10mg/kg提高至180mg/kg时,其总体清除率提高了5倍,但根据血浆中游离药物浓度计算出的清除率则不随剂量变化。作者推断该现象是由于药物与血浆蛋白的非线性结合所致。

38
五、药物的非线性排泄研究

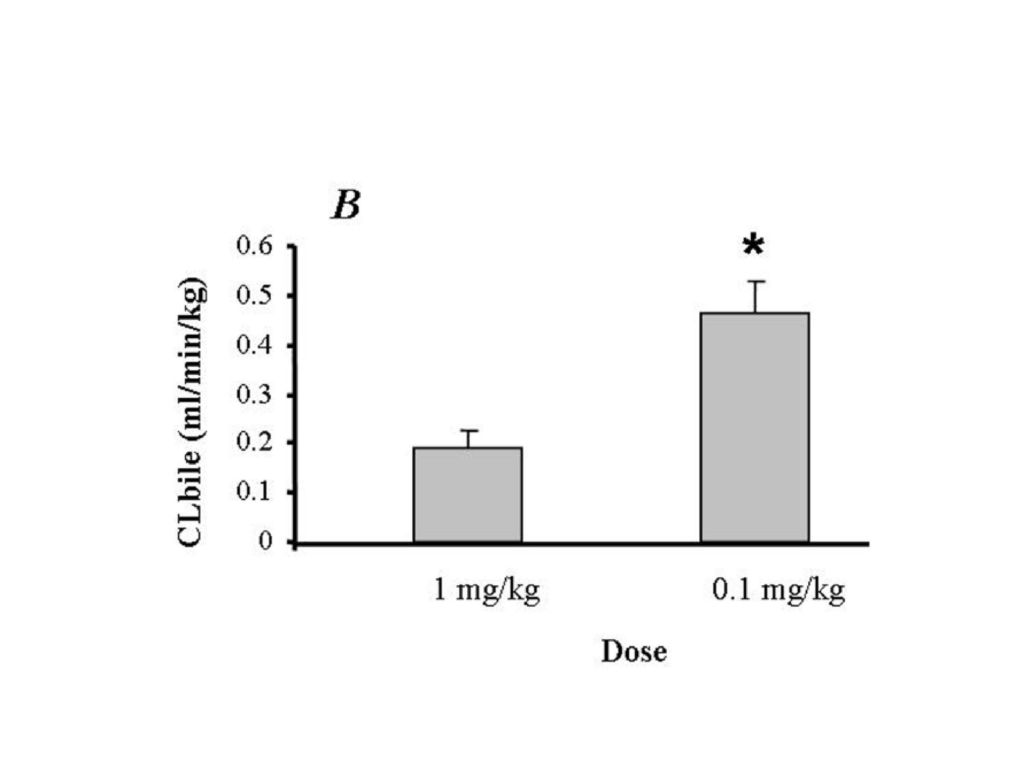
44
思考题: 1.什么是非线性药物动力学? 2.若某药物存在非线性消除现象,如何设计一个试验予以证实? 3.对于非线性消除的药物,试分别列出口服、静注和静滴给药后血药浓度变化的速度方程 dC/dt = ?

Similar presentations









班 毕业时间:2012年6月.>")













