Download presentation
Presentation is loading. Please wait.

1
五、蛋白质药物质量控制的要点

2
1、蛋白质纯度检查 2、蛋白质含量测定 3、蛋白质药物理化性质的鉴定 4、残余杂质检测

3
1、蛋白质纯度检查 世界卫生组织规定必须用HPLC和非还原SDS-PAGE两种方法测定,其纯度都应达到95%以上才能合格。
某些重组药物的纯度要求更高,要达到99%以上。 纯度的检测通常是在原液(drug substance,bulk)中进行。
中进行。")
4
如何认识蛋白质类药物纯度检测? 1 从蛋白质制剂中检测出少量的污染蛋白质是很困难的。因为污染蛋白质的量可能低于很多测定方法的检测下限。制剂中往往含有大量的辅料。 2 当用一种方法测定蛋白质纯度时,可能有两种或更多的蛋白质表现出相似的行为。这种类似的行为可能会导致本来是混合物的样品也被认为是均一物质的错误结论。

5
3 只用一种方法作为纯度试验的标准是很不可靠的,必须选择多种测定纯度的方法。
3 只用一种方法作为纯度试验的标准是很不可靠的,必须选择多种测定纯度的方法。 最好的纯度标准是建立多种分析方法,从等电点、相对分子质量、疏水性等不同的角度来证明了蛋白质样品的均一性。

6
5 没有一个真正的检验纯度的方法,只有检测样品不纯或非均一的方法。
4 纯度最终取决于所用方法的类型和分辨力,低分辨率方法检测合格的样品改用高分辨力方法时就有可能证明它是不纯的。 5 没有一个真正的检验纯度的方法,只有检测样品不纯或非均一的方法。

7
1.1非还原SDS-PAGE电泳法 银染法染色,加样量不低于5μg。 考马斯亮蓝R-250染色,加样量不低于10μg。 结果应无明显杂蛋白出现,经扫描仪扫描,蛋白量应不低于总蛋白量的95%或98%。

8
1.2 HPLC法(分子筛层析、反相HPLC、离子交换层析等)
HPLC法应根据不同的纯化工艺选择不同的方法。一般尽量采用与SDS- PAGE法原理不同的反相柱或其他离子交换柱进行分析,而不主张用分子筛分析。在质量标准中要说明采用的是什么性质的分析柱。如有些产品不适合用反相柱,要说明原因。

9
毛细管电泳的方法简便、快速,灵敏度和分辨率高,但价格昂贵,重现性差,尚未作为常规检定。
1.3 毛细管电泳 毛细管电泳的方法简便、快速,灵敏度和分辨率高,但价格昂贵,重现性差,尚未作为常规检定。

12
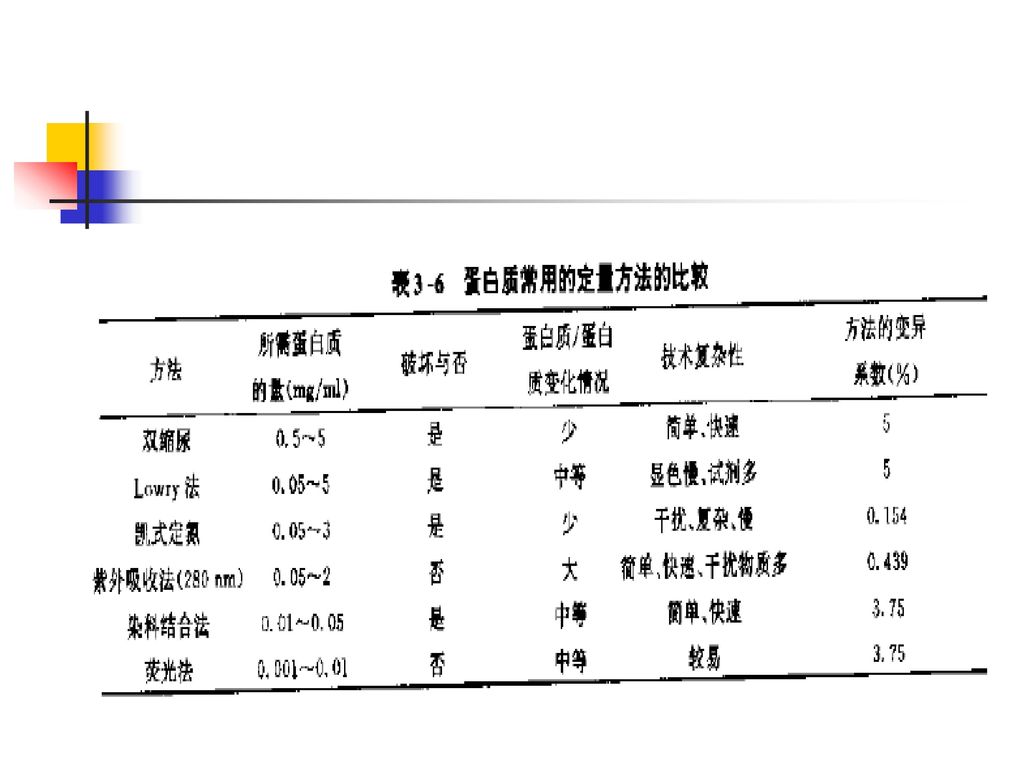
2、蛋白质含量测定 在质量标准中设定此项目主要用于原液比活性计算和成品(drug product)规格的控制。 蛋白含量采用Folin-酚试剂法(Lowry法)、染色法(Bradford法)、双缩脲法、紫外吸收法、HPLC法和凯氏定氮等方法。 其中Lowry法和Bradford法是在质量检定中经常使用的方法。

13
2.1凯氏定氮法 不需要昂贵的仪器设备,操作比较繁琐,灵敏度较低(毫克水平),其结果受非蛋白氮的影响,可用钨酸沉淀法或三氯醋酸沉淀法加以排除。
,其结果受非蛋白氮的影响,可用钨酸沉淀法或三氯醋酸沉淀法加以排除。")
14
2.2 Folin-酚试剂法(Lowry法) 灵敏范围:5~100μg/ml,该方法适合蛋白质的微量测定。 优点:方法简便,灵敏度较高,较紫外吸收法灵敏10~20倍,较双缩脲法灵敏100倍左右。所用仪器简单,不同蛋白质间的变异少,是蛋白质量化的可靠方法。所需时间:约40min。目前国内外多数重组制品原液蛋白含量用该法进行测定。

15
缺点: 受多种物质干扰,反应速度慢,某些试剂不稳定,蛋自质不可逆变性,芳香族氨基酸比例会影响蛋白检测结果。

16
2.3 BCA法 从Lowry法派生的蛋白含量测定方法。 优点:操作比较简单,采用单一试剂4,4’-二羧酸-2,2’-二喹啉(bicichoninic Acid,BCA),终产物稳定,比Lowry法的干扰物少;所需时间:2h或过夜。 缺点:反应时间长,蛋白质不可逆变性。灵敏范围:标准分析:10~1200μg/mi;微量分析:0.5~10μg/ml。

17
2.4 Bradford法 Bradford法是采用考马斯亮蓝G-250与蛋白质结合的原理,迅速、敏感的定量测定蛋白质的方法。需要时间;10min。灵敏范围:25~200μg/ml;0.1ml的最小体积可测得的最低蛋白为2.5μg。

18
该方法与Lowry法比较有更加简便、快速 (2min的显色时间)、灵敏,只需少量蛋白,干扰物质少等优点。缺点是不同的纯化蛋白间有可变性,该方法使蛋白不可逆变性。
国外已有重组制品将Bradford法作为蛋白含量常规检定方法。

19
2.5紫外吸收法 灵敏范围:0.2~2mg/m1;微管中最低可检出0.1m1(0. 05mg)。 需要的时间:几分钟。方法简单,样品可回收,但结果受光吸收物质影响。 适合于含有共轭双键较多的芳香族氨基酸的蛋白质含量测定。
。 需要的时间:几分钟。方法简单,样品可回收,但结果受光吸收物质影响。 适合于含有共轭双键较多的芳香族氨基酸的蛋白质含量测定。 .")
20
该测定方法简单、灵敏、快速,在蛋白质和酶的生化制备中广泛应用,特别是在柱层析分离中,利用280nm进行紫外检测来判断蛋白质吸附或洗脱情况是最常用的方法。如重组尿激酶原(Pro-UK)的质量检测标准中的蛋白含量测定项目就采用此法完成。 优点:快速,非破坏性,直接测定不需要标准品,不消耗样品,低浓度的盐类不干扰测定。 缺点①如果一种蛋白不含苯丙氨酸、酪氨酸和色氨酸,它将无法检测;②若样品中含有嘌呤、嘧啶等吸收紫外光的物质,会出现较大干扰。

21
2.6 ELISA法 为特异蛋白含量测定方法,具有特异性强,灵敏度高,同时测定样品多等特点,可用于生产过程中目标蛋白含量的测定。

22
2.7 HPLC法 具有定量精确,灵敏度高,重复性好,自动化程度高等特点。 需要同种蛋白做标准品。

23
2.8 SDS-PAGE-CBB G250(Coomassie brilliant blue G-250,CBBG ) 染色-比色法
结合Lowry法等总蛋白含量的测定,可用于复杂的蛋白混合物中目标蛋白的定量测定。

24
重组制品原液蛋白量少,纯度高,可用Lowry法或Bradford法测定蛋白含量。
细胞因子蛋白含量正逐步采用HPLC法,HPLC法测定蛋白含量能够排除溶剂系统的干扰,特别适用于进行批与批之间的质量控制。

26
3、蛋白质药物理化性质的鉴定 3.1 特异性鉴别试验 主要确定蛋白质的抗原性 免疫学方法:根据抗原抗体特异反应建立的方法。
重组产品通常用免疫印迹和点免疫进行鉴定,特别当电泳出现两条或两条以上区带时则应该用免疫印迹进行鉴定。 半干胶转移免疫印迹法是最常用的方法。

27
印迹的效率取决于蛋白质从胶上转移到膜上的效率以及蛋白质与膜结合的效率。在印迹实验开始时,蛋白质位于凝胶内部并带有十二烷基硫酸钠(SDS)。SDS的负电性使得SDS蛋白复合物移向正极。在移动过程中,SDS不断地从蛋白质上脱离。当蛋白质到达疏水膜,一些疏水结合位点不带有SDS时,可以与膜发生疏水互相作用。蛋白质结合到膜上,SDS不断地从蛋白质脱离并移向正极而蛋白质则留在了膜上。
。SDS的负电性使得SDS蛋白复合物移向正极。在移动过程中,SDS不断地从蛋白质上脱离。当蛋白质到达疏水膜,一些疏水结合位点不带有SDS时,可以与膜发生疏水互相作用。蛋白质结合到膜上,SDS不断地从蛋白质脱离并移向正极而蛋白质则留在了膜上。")
28
3.2 相对分子质量测定 还原型SDS-PAGE法测定 在Mr为15 000~ 的范围,与其他方法测得的Mr相比,误差一般在±10%以内, 凝胶过滤(分子筛)法:根据蛋白分子大小和形状进行测定,误差比SDS-PAGE大,目前应用较少。
法:根据蛋白分子大小和形状进行测定,误差比SDS-PAGE大,目前应用较少。 .")
29
质谱法: 具有准确、快速,重复性好,测定范围广等特点,当相对分子质量小于10 000的蛋白质用上述方法因误差太大而无法测定其相对分子质量时,可用该法进行蛋白质相对分子质量的确认实验如重组人EGF、重组人bFGF和重组水蛭素等。

30
3.3等电点测定 可以表征药物的理化性质和纯度 均一的重组蛋白质只有一个等电点,有时因加工 修饰等影响可出现多个等电点,但应有一定的范 围。所以等电点测定是控制重组产品生产工艺稳 定性的重要指标。

31
常用的测定方法: 等电聚焦电泳法(IEF): 毛细管电泳法(CE):该法用紫外检测,适用于某些不 易染色的蛋白质多肽,如EGF、hCGRP等制品的测 定。 标准中等电点的规定: 等电点标准应规定为在±0.5pH范围之内,应有明 显的主显带。

32
3.4肽图分析 概念: 意义:一级结构 ,工艺稳定性 肽图的裂解方法 : (1)化学裂解法 (2)蛋白酶裂解法 (3)特殊处理: 二硫键、空间构象较复杂的特殊产品
化学裂解法 (2)蛋白酶裂解法 (3)特殊处理: 二硫键、空间构象较复杂的特殊产品")
33
肽图常用的分析方法有: (1)SDS-PAGE电泳: (2)反相HPLC(RP-HPLC): (3)毛细管电泳: (4)液质分析:
SDS-PAGE电泳: (2)反相HPLC(RP-HPLC): (3)毛细管电泳: (4)液质分析:")
35
3.5 吸收光谱 某一重组蛋白质来说,其最大吸收波长是固定的; 在生产过程中每批产品的紫外吸收光谱应当是一致的。 有的重组产品一级结构不含芳香族氨基酸,在280nm 附近没有最大吸收峰,可不做紫外吸收光谱测定, 如重组脑利钠肽(rhBNP)。
。")
36
3.6 氨基酸组成分析 是结构分析的辅助手段,质量控制的重要指标. 在生产的控制中,样品的氨基酸组成结果应与标 准品一致。 这在试生产的头三批或工艺改变时应当测定。

37
3.7 N末端和C末端氨基酸测序 作为重组蛋白质和肽的重要鉴别指标,一般要 求至少测定N端15个氨基酸。在中试头三批产品 应当测定;C端定1~3个,但在我国现有法规中 C端不一定要测定。

38
3.8 蛋白质的二硫键分析 二硫键和巯基与蛋白质的生物活性密切相关。测 定巯基的方法有PMCB法、DTNB法和NEMI法等。对 二硫键的分析虽然在常检定项目中没有规定,但在质 量研究中应尽可能分析清楚。 有些产品二硫键较多,用现有的技术完全分析清楚很 困难,可以结合其他项目的检测,如比活性等进行有 效的质量控制。

39
举例:IL-2分子中有Cys-58和Cys-105形成的二硫键,Cys-125是以游离巯基形式存在。
错误配对,生物活性原来的1/400,会引起抗原性增强。 分析:pH8.5下用3H-碘乙酸处理IL-2,使游离巯基与3H-碘乙酸作用形成衍生物。然后再还原打开二硫键,并用14C-碘乙酸处理,结果3H-标记仅在肽-11上(Cys-125),而肽-11基本上无14C放射性。相反14C -的放射性全在肽-12和肽-14上(Cys-58与Cys-105的二硫键位置上)。
,而肽-11基本上无14C放射性。相反14C -的放射性全在肽-12和肽-14上(Cys-58与Cys-105的二硫键位置上)。")
40
3.9 糖蛋白中糖的分析 对糖蛋白化学结构分析的意义: 附着于蛋白质的寡糖可以上调、下调或抑制糖蛋白的生物活性,还可以影响蛋白质的代谢命运、稳定性或溶解性. 通过基因工程方法将一种糖蛋白在不同细胞中表达,可以导致生产不同类型的糖蛋白。即使在相同的细胞类型中,也可能会出现糖形成时由于一些寡糖的精细结构不同和普通寡糖产生的相对频率不同而产生截然不同的糖蛋白。

41
糖蛋白的糖基化分析 : 糖基化位点,分子量,糖的组成,连接方式, 分析方法: 酶解,质谱,LC/MS,GC/MS

42
4、残余杂质检测 残余杂质可分为外来污染物和与产品相关的杂质两大类。
外来污染物包括:微生物污染、热原、细胞的成分(例如细胞的蛋白质,DNA其他的组分)、培养基中的成分、来自生产过程各步骤中的物质、来自产品纯化步骤中的物质(亲和柱中的抗体,其他试剂);
、培养基中的成分、来自生产过程各步骤中的物质、来自产品纯化步骤中的物质(亲和柱中的抗体,其他试剂);")
43
与产品相关的杂质包括: 突变物、错误裂解的产品、二硫化物异构体、二聚体和多聚体;化学修饰的形态:脱去酰氨基的或氧化的形态、其他降解产物等。

44
4.1宿主细胞蛋白含量 概念: 宿主细胞蛋白质一般简称宿主蛋白,是指生产过程中来自宿主或培养基的残留肽等杂质。基因工程药物中的宿主蛋白含量,可用 ELISA法(enzyme-linked immunosorbent assay)测定。 在临床使用中需要反复多次注射的药品,则需要做残余菌体蛋白含量测定,所用方法以双抗体夹心ELISA为宜.

45
测定方法 1包被: 在96孔细胞培养板中,以兔抗大肠杆菌肽组分(periplasmic E.coli peptides,简称PECP)抗体包被,4℃放置过夜(16~18小时),用洗涤液洗板3次. 2加样: 加入样品及PECP对照标准培养,37℃放置2小时.

46
3加抗体: 经洗涤后,加入抗PECP/HRP结合物(anti-PECP/HRP conjugate)[HRP为辣根过氧化物酶(horseradish peroxidase)的简称]使其与固相抗体结合。 4检测: 结合的酶量用过氧化氢和四甲基联苯胺(3,3’5,5’tetra methylbenzidine,简称TMBZ)为底物进行测定。最后氧化的黄色产物于450nm波长处测定其吸收度,从而求出样品中宿主蛋白的含量。
![3加抗体: 经洗涤后,加入抗PECP/HRP结合物(anti-PECP/HRP conjugate)[HRP为辣根过氧化物酶(horseradish peroxidase)的简称]使其与固相抗体结合。](http://slidesplayer.com/slide/11365153/61/images/46/%EF%BC%93%E5%8A%A0%E6%8A%97%E4%BD%93%EF%BC%9A+%E7%BB%8F%E6%B4%97%E6%B6%A4%E5%90%8E%EF%BC%8C%E5%8A%A0%E5%85%A5%E6%8A%97PECP%2FHRP%E7%BB%93%E5%90%88%E7%89%A9%28anti-PECP%2FHRP+conjugate%29%5BHRP%E4%B8%BA%E8%BE%A3%E6%A0%B9%E8%BF%87%E6%B0%A7%E5%8C%96%E7%89%A9%E9%85%B6%28horseradish+peroxidase%29%E7%9A%84%E7%AE%80%E7%A7%B0%5D%E4%BD%BF%E5%85%B6%E4%B8%8E%E5%9B%BA%E7%9B%B8%E6%8A%97%E4%BD%93%E7%BB%93%E5%90%88%E3%80%82.jpg "4检测: 结合的酶量用过氧化氢和四甲基联苯胺(3,3’5,5’tetra methylbenzidine,简称TMBZ)为底物进行测定。最后氧化的黄色产物于450nm波长处测定其吸收度,从而求出样品中宿主蛋白的含量。")
47
4.2 宿主细胞DNA残留量测定方法 概念: 在利用工程菌生产蛋白质药物时,工程菌的DNA可能会在蛋白质药物终产品中有一定的残留,这些DNA可能会对药物的安全性和有效性有影响,应测定和控制其含量。

48
限量要求: 世界卫生组织(WHO)要求每一剂量药物中残余DNA含量不得超过100pg。 我国《人用重组DNA制品质量控制要点》小于100pg/剂是安全的。 基因工程药物中残余DNA的含量限度一般定为每一剂量<100pg。
要求每一剂量药物中残余DNA含量不得超过100pg。 我国《人用重组DNA制品质量控制要点》小于100pg/剂是安全的。 基因工程药物中残余DNA的含量限度一般定为每一剂量<100pg。")
49
测定原理: 供试品中的DNA经变性为单链后吸附于固相膜上,在一定温度下,可与相匹配的单链DNA复性而重新结合成为双链DNA,称为杂交。将特异性DNA探针标记后,与吸附在固相膜上的供试品单链DNA杂交,并使用与标记物相应的显示系统显示杂交结果,与已知含量的阳性DNA对照对比后,可测定供试品中外源性DNA的含量。

50
测定方法 1.标记 DNA在随机启动标记前须经热变性。标记反应快速(1h),其结果使新合成的DNA中每20~25个核昔酸掺入一个地高辛苷配基。(用于探针标记和阳性对照的DNA,由生产供试品用的传代细胞、工程菌或杂交瘤细胞提取纯化获得) 2.杂交 半抗原标记的DNA与硝酸纤维素膜或尼龙膜上DNA的杂交。

51
3.免疫检测 杂交后的滤膜上的半抗原标记DNA与抗体复合物(抗地高辛苷配基-碱性磷酸酶复合物)结合。酶促反应在碱性pH时进行,此时需加入无色的BCIP( 5-溴-4-氯-3-吲哚磷酸(5-bromo-4-chloro-3-indolyl phosphate)和NBT(氮蓝四唑)。数分钟后开始生成蓝色沉淀,并继续生成,直至3d。通常反应可以在24h后终止。
结合。酶促反应在碱性pH时进行,此时需加入无色的BCIP( 5-溴-4-氯-3-吲哚磷酸(5-bromo-4-chloro-3-indolyl phosphate)和NBT(氮蓝四唑)。数分钟后开始生成蓝色沉淀,并继续生成,直至3d。通常反应可以在24h后终止。")
52
4.3 鼠源型IgG含量 如在纯化过程中用到单克隆抗体亲和柱,则半成品应有鼠IgG含量检测;在采用鼠单克隆抗体进行纯化时,必须测定IgG残留量。一般认为每剂量应小于10ng是安全的。

53
4.4 蛋白A含量 如所用亲和柱含有蛋白A(protein A),必须设定蛋白A的测定项目,采用ELISA方法测定原液中的残留量应不高于规定值。
,必须设定蛋白A的测定项目,采用ELISA方法测定原液中的残留量应不高于规定值。")
54
4.5小牛血清残留量 在生产过程中如加有小牛血清,必须测定其残留量。采用2005年版《中国生物制品规程》所规定的方法测定。

55
4.6 残余抗生素 原则上不主张使用抗生素。如果在生产工艺中使用了抗生素,不仅要在纯化工艺中除去,而且要对终产品进行检测。
用生物法测定残余量,阳性对照管周围应出现光滑、清晰的抑菌圈并与所含阳性对照呈显著的量效关系。阴性对照管周围应无抑菌圈出现。待检样品结果阴性者判为合格。 另外在检测中要注意的是,如果制品中含SDS,对残余抗生索活性测定有干扰作用。

56
4.7 内毒素含量(LAL) 按《中国药典》规定的方法测定,采用固定厂家的鲎试剂盒,均为保证测定的准确性的必要条件。
 按《中国药典》规定的方法测定,采用固定厂家的鲎试剂盒,均为保证测定的准确性的必要条件。")
57
4.8 产品相关物质 在与产品有关的物质中,有许多已知的与天然组分具有相同的生物活性的组分,例如胰岛素的脱酰氨基衍生物或人生长激素的脱酰氨基和亚砜衍生物。它们可能被认为是活性成分,但应制定允许的限度。

58
4.9 其他杂质 主要包括生产和纯化过程中加入的其他物质如铜、锌离子,抗生素,甲醛,SDS等杂质检测 . 通过清除或者限制已知杂质的数量,来避免这些物质所带来的特殊危险。

59
检测方法 微量的生物大分子:酶联免疫学或类似的方法进行定量或限量分析 小分子物质:液相色谱,气相色谱等分析化学方法或敏感的生物学方法。 具有自我复制、繁殖能力的病毒、支原体:生物学、分子生物学、生物化学和免疫学等方法 检测方法进行方法学验证 建立标准化的操作程序

60
蛋 白 质 药 物 质 量控 制 举 例

61
一、人用重组DNA制品 1 人用重组DNA制品:
由于分子遗传学、核酸化学及重组DNA(rDNA)技术的迅速发展,现已能够确定和获得许多天然活性蛋白的编码基因,将其插入表达载体或引入某种宿主细胞后,能有效地表达该基因产物,再经分离、纯化和检定,可得到用于预防和治疗某些人类疾病的制品,诸如现有的乙型肝炎疫苗、胰岛素、生长激素、干扰素等。
技术的迅速发展,现已能够确定和获得许多天然活性蛋白的编码基因,将其插入表达载体或引入某种宿主细胞后,能有效地表达该基因产物,再经分离、纯化和检定,可得到用于预防和治疗某些人类疾病的制品,诸如现有的乙型肝炎疫苗、胰岛素、生长激素、干扰素等。")
62
2、质量控制要求

63
(一 )原材料的控制 1.表达载体和宿主细胞 应提供有关表达载体详细资料,包括基因的来源、克隆和鉴定,表达载体的构建、结构和遗传特性。
应说明载体组成各部分的来源和功能,如复制子和启动子来源,或抗生素抗性标志物。提供至少包括构建中所用位点的酶切图谱。

64
应提供宿主细胞的资料,包括细胞株(系)名称、来源、传代历史、检定结果及基本生物学特性等。
应详细说明载体引入宿主细胞的方法及载体在宿主细胞内的状态(是否整合到染色体内)及拷贝数。应提供宿主和载体结合后的遗传稳定性资料。
及拷贝数。应提供宿主和载体结合后的遗传稳定性资料。")
65
2.克隆基因的序列 应提供插入基因和表达载体两侧端控制区的核苷酸序列。所有与表达有关的序列均应详细叙述。 3.表达 应详细叙述在生产过程中,启动和控制克隆基因在宿主细胞中的表达所采用的方法及表达水平。 4.原辅料 原辅料应按照国家药品监督管理局有关规定执行。动物源性原料的使用应提供来源及质控检测资料;发酵用培养基不能添加β内酰胺类抗生素。

66
1.主细胞库(MASTERCELLBANK)
(二)生产的控制 1.主细胞库(MASTERCELLBANK) 2.有限代次生产 3.连续培养生产 4.纯化
生产的控制. 1.主细胞库(MASTERCELLBANK) 2.有限代次生产. 3.连续培养生产. 4.纯化.")
67
(三)最终产品的控制 1.物理化学鉴定 (1)氨基酸组成 (2)氨基酸末端序列 N-端和C-端氨基酸 (3)肽谱 (4)巯基和二硫键
巯基和/或二硫键的数量和位置。 (5)碳水化合物结构 中性糖、氨基糖、唾液酸
碳水化合物结构. 中性糖、氨基糖、唾液酸.")
68
(6)分子量 应用分子筛层析法、SDS-PAGE(还原和/或非还原条件下)、质谱测定法。 (7)等电点 通过等电聚焦电泳或其他适当的方法测定。 (8)消光系数(或克分子吸光度) (9)电泳图型 (10)液相层析图谱 分子筛层析、反相液相层析、离子交换液相层析、亲和层析获得目的产品/药物的一致性、同一性和纯度的层析图谱和数据。 (11)光谱分析
液相层析图谱. 分子筛层析、反相液相层析、离子交换液相层析、亲和层析获得目的产品/药物的一致性、同一性和纯度的层析图谱和数据。 (11)光谱分析.")
69
2.杂质检查 (1)工艺相关杂质 工艺相关杂质来源于生产工艺,可分三大类:来源于细胞基质、培养基和下游工艺。
①来源于细胞基质的杂质包括源于宿主生物体的蛋白/多肽;核酸(宿主细胞/载体/总DNA);多糖及病毒。 ②来源于培养基的杂质包括诱导剂(多核苷酸,病毒)、抗生素、血清及其他培养基组分。 ③来源于下游工艺产生的杂质包括酶、化学/生化处理试剂(如溴化氰、胍、氧化剂和还原剂)、无机盐(如重金属、砷、非有色金属离子)、溶剂、载体/配体(如单克隆抗体),及其他可滤过的物质。
;多糖及病毒。 ②来源于培养基的杂质包括诱导剂(多核苷酸,病毒)、抗生素、血清及其他培养基组分。 ③来源于下游工艺产生的杂质包括酶、化学/生化处理试剂(如溴化氰、胍、氧化剂和还原剂)、无机盐(如重金属、砷、非有色金属离子)、溶剂、载体/配体(如单克隆抗体),及其他可滤过的物质。")
70
(2)产品相关杂质 ①化学修饰类型:应考虑脱酰胺、异构化、错配S-S连接和氧化形式的分离和鉴别。 ②降解物和聚合体:聚合体包括二聚体和多聚体:可用分子筛层析法(如SE-HPLC)进行定量;降解物:应建立降解物的判定标准,并对稳定性试验产生的降解产物进行监测。
产品相关杂质 ①化学修饰类型:应考虑脱酰胺、异构化、错配S-S连接和氧化形式的分离和鉴别。 ②降解物和聚合体:聚合体包括二聚体和多聚体:可用分子筛层析法(如SE-HPLC)进行定量;降解物:应建立降解物的判定标准,并对稳定性试验产生的降解产物进行监测。")
71
3.生物学测定 应用免疫印迹法 (2)效价测定 采用国际或国家参考品,以体内或体外法测定制品的生物学活性,并标明其活性单位。
(1)鉴别试验 应用免疫印迹法 (2)效价测定 采用国际或国家参考品,以体内或体外法测定制品的生物学活性,并标明其活性单位。 (3)特异比活性测定 测定其特异比活性,以活性单位/重量表示。 (4)热原质试验
鉴别试验. 应用免疫印迹法. (2)效价测定. 采用国际或国家参考品,以体内或体外法测定制品的生物学活性,并标明其活性单位。 (3)特异比活性测定. 测定其特异比活性,以活性单位/重量表示。 (4)热原质试验.")
72
(5)无菌试验 (6)抗原性物质检查 抗原可能产生的抗体或变态反应。 (7)异常毒性试验
无菌试验 (6)抗原性物质检查 抗原可能产生的抗体或变态反应。 (7)异常毒性试验")
73
本品系由含有高效表达人干扰素α2b基因的大肠杆菌,经发酵、分离和高度纯化后制成。含适宜稳定剂,不含防腐剂和抗生素。
拼音名:Chongzu Ren Ganraosu α2b Zhusheye 英文名:Recombinant Human Interferon α2b Injection 本品系由含有高效表达人干扰素α2b基因的大肠杆菌,经发酵、分离和高度纯化后制成。含适宜稳定剂,不含防腐剂和抗生素。

74
1 基本要求 生产和检定用设施、原料及辅料、水、器具、动物等应符合“凡例”的有关要求。

75
2 制造 2.1 工程菌菌种 2.1.1 名称及来源 重组人干扰素α2b工程菌株系由带有人干扰素α2b基因的重组质粒转化的大肠杆菌菌株。 2.1.2 种子批的建立 2.1.3 菌种检定 主种子批和工作种子批的菌种应进行以下各项全面检定。 2.1.3.1 划种LB琼脂平板 应呈典型大肠杆菌集落形态,无其他杂菌生长。 2.1.3.2 染色镜检 应为典型的革兰阴性杆菌。

76
2.1.3.3 对抗生素的抗性应与原始菌 2.1.3.4 电镜检查(工作种子批可免做) 应为典型大肠杆菌形态,无支原体、病毒样颗粒及其他微生物污染。 2.1.3.5 生化反应 应符合大肠杆菌生物学性状。 2.1.3.6 干扰素表达量 在摇床中培养,应不低于原始菌种的表达量。 2.1.3.7 表达的干扰素型别 应用抗α2b型干扰素参考血清做中和试验,证明型别无误。 2.1.3.8 质粒检查 该质粒的酶切图谱应与原始重组质粒相符。

77
将检定合格的工作种子批菌种接种于适宜的培养基(可含适量抗生素)中培养。 2.2.2 发酵用培养基 采用适宜的不含任何抗生素的培养基。
2.2 原液 2.2.1 种子液制备 将检定合格的工作种子批菌种接种于适宜的培养基(可含适量抗生素)中培养。 2.2.2 发酵用培养基 采用适宜的不含任何抗生素的培养基。 2.2.3 种子液接种及发酵培养 2.2.3.1 在灭菌培养基中接种适量种子液。 2.2.3.2 在适宜的温度下进行发酵,应根据经批准的发酵工艺进行,并确定相应的发酵条件,如温度、pH值、溶氧、补料、发酵时间等。发酵液应定期进行质粒丢失率检查。
中培养。 2.2.2 发酵用培养基. 采用适宜的不含任何抗生素的培养基。 2.2.3 种子液接种及发酵培养. 2.2.3.1 在灭菌培养基中接种适量种子液。 2.2.3.2 在适宜的温度下进行发酵,应根据经批准的发酵工艺进行,并确定相应的发酵条件,如温度、pH值、溶氧、补料、发酵时间等。发酵液应定期进行质粒丢失率检查。")
78
2.2.4 发酵液处理 用适宜的方法收集处理菌体。 2.2.5 初步纯化 2.2.6 高度纯化 经初步纯化后,采用经批准的工艺进行高度纯化,使其达到3.1项要求,即为干扰素原液。加入适宜稳定剂,除菌过滤后保存于适宜温度,并规定其有效期。

79
2.2.7 原液检定 按3.1项进行。

80
2.3 半成品 2.3.1 配制与除菌 2.3.1.1 稀释液配制 按经批准的配方配制稀释液。配制后应立即用于稀释。 2.3.1.2 稀释与除菌 将检定合格加稳定剂的干扰素原液用 项稀释液稀释至所需度,除菌过滤后即为半成品,保存于2~8℃。 2.3.2 半成品检定 按3.2项进行。

81
2.4 成品 2.4.1 分批 应符合“生物制品分批规程”规定。 2.4.2 分装 应符合“生物制品分装和冻干规程”规定。 2.4.3 规格 应为经批准的规格。 2.4.4 包装 应符合“生物制品包装规程”规定。

82
3 检定 3.1 原液检定 3.1.1 生物学活性 依法测定(附录Ⅹ C)。 3.1.2 蛋白质含量 依法测定(附录Ⅵ B第二法)。 3.1.3 比活性 为生物学活性与蛋白质含量之比,每1mg蛋白质应不低于1.0×10〈8〉IU。
。 3.1.3 比活性. 为生物学活性与蛋白质含量之比,每1mg蛋白质应不低于1.0×10〈8〉IU。 .")
83
3.1.4 纯度 3.1.4.1 电泳法 依法测定(附录Ⅳ C)。用非还原型SDS-聚丙烯酰胺凝胶电泳法,分离胶胶浓度为15%,加样量应不低于l0μg(考马斯亮蓝R250染色法)或5μg(银染法)。经扫描仪扫描,纯度应不低于95.0%。 3.1.4.2 高效液相色谱法 依法测定(附录Ⅲ B)。色谱柱以适合分离分子质量为5~60kD蛋白质的色谱用凝胶为填充剂;流动相为0.1mol/L磷酸盐-0.1mol/L氯化钠缓冲液,pH7.0;上样量不低于20μg,于波长280nm处检测,以干扰素色谱峰计算理论板数应不低于1000。按面积归一化法计算,干扰素主峰面积应不低于总面积的95.0%。
。色谱柱以适合分离分子质量为5~60kD蛋白质的色谱用凝胶为填充剂;流动相为0.1mol/L磷酸盐-0.1mol/L氯化钠缓冲液,pH7.0;上样量不低于20μg,于波长280nm处检测,以干扰素色谱峰计算理论板数应不低于1000。按面积归一化法计算,干扰素主峰面积应不低于总面积的95.0%。")
84
3.1.5 分子量 依法测定(附录Ⅳ C)。用还原型SDS-聚丙烯酰胺凝胶电泳法,分离胶胶浓度为15%,加样量应不低于1.0μg,制品的分子质量应为19.2kD±1.92kD。 3.1.6 外源性DNA残留量 每1次人用剂量应不高于10ng(附录Ⅸ B)。 3.1.7 鼠IgG残留量 如采用单克隆抗体亲和色谱法纯化,应进行本项检定。每1次人用剂量鼠IgG残留量应不高于100ng。
。 3.1.7 鼠IgG残留量. 如采用单克隆抗体亲和色谱法纯化,应进行本项检定。每1次人用剂量鼠IgG残留量应不高于100ng。")
85
依法测定(附录Ⅸ A),不应有残余氨苄西林或其他抗生素活性。 3.1.10 细菌内毒素检查
3.1.8 宿主菌蛋白残留量 应不高于总蛋白质的0.10%(附录Ⅸ C)。 3.1.9 残余抗生素活性 依法测定(附录Ⅸ A),不应有残余氨苄西林或其他抗生素活性。 3.1.10 细菌内毒素检查 每300万IU应小于10EU(附录Ⅻ E凝胶限量试验)。 3.1.11 等电点 主区带应为4.0~6.7(附录Ⅳ D)。 3.1.12 紫外光谱扫描 最大吸收峰波长应为278nm±3nm(附录Ⅱ A)。
。 3.1.9 残余抗生素活性. 依法测定(附录Ⅸ A),不应有残余氨苄西林或其他抗生素活性。 3.1.10 细菌内毒素检查. 每300万IU应小于10EU(附录Ⅻ E凝胶限量试验)。 3.1.11 等电点. 主区带应为4.0~6.7(附录Ⅳ D)。 3.1.12 紫外光谱扫描. 最大吸收峰波长应为278nm±3nm(附录Ⅱ A)。")
86
3.1.13 肽图(至少每半年测定1次) 依法测定(附录Ⅷ E),应与对照品图形一致。 3.1.14 N-末端氨基酸序列(至少每年测定1次) 用氨基酸序列分析仪测定,N-末端序列应为: Cys-Asp-Leu-Pro-Gln-Thr-His-Ser-Leu-Gly-Ser-Arg-Arg-Thr-Leu。

87
3.2 半成品检定 3.2.1 细菌内毒素检查 每300万IU应小于10EU(附录Ⅻ E凝胶限量试验)。 3.2.2 无菌检查 依法检查(附录Ⅻ A),应符合规定。 3.3 成品检定 3.3.1 鉴别试验 按免疫印迹法(附录Ⅷ A)或免疫斑点法(附录Ⅷ B)测定,应为阳性。
或免疫斑点法(附录Ⅷ B)测定,应为阳性。")
88
按附录ⅠA中装量项进行。应不低于标示量。 3.3.3 pH值 应为6.5~7.5(附录Ⅴ A)。 3.3.4 生物学活性
3.3.2 物理检查 3.3.2.1 外观 应为澄明液体。 3.3.2.2 可见异物 依法检查(附录Ⅴ B),应符合规定。 3.3.2.3 装量 按附录ⅠA中装量项进行。应不低于标示量。 3.3.3 pH值 应为6.5~7.5(附录Ⅴ A)。 3.3.4 生物学活性 应为标示量的80%~150%(附录Ⅹ C)。
,应符合规定。 3.3.2.3 装量. 按附录ⅠA中装量项进行。应不低于标示量。 3.3.3 pH值. 应为6.5~7.5(附录Ⅴ A)。 3.3.4 生物学活性. 应为标示量的80%~150%(附录Ⅹ C)。")
89
3.3.5 无菌检查 依法检查(附录Ⅻ A),应符合规定。 3.3.6 细菌内毒素检查 每1次人用剂量应小于10EU(附录Ⅻ E凝胶限量试验)。 3.3.7 异常毒性检查 依法检查(附录Ⅻ F小鼠试验法),应符合规定。 4 保存、运输及有效期 于2~8℃避光保存和运输。自分装之日起,按批准的有效期执行。 5 使用说明 应符合“生物制品包装规程”规定。

Similar presentations





:核蛋白体组成成分 转移 RNA ( tRNA ):转运氨基酸 信使 RNA ( mRNA ):蛋白质合成模板 不均一核 RNA ( hnRNA ):成熟 mRNA 的前体 小核 RNA ( snRNA ):>")






(P298)>")



受体拮抗剂 抑制性氨基酸受体受体拮抗剂 神经肽Y受体拮抗剂>")




