Download presentation
1
有机化学反应历程 若按底物键的断裂方式分类: 若按反应物与产物之间的关系分类: 常常有些反应是几种反应类型的综合:
1.离子反应或异裂反应; 2.自由基反应或均裂反应; 3.协同反应。 若按反应物与产物之间的关系分类: 亲电取代、亲核取代、自由基取代;亲电加成、亲核加成、自由基加成、协同加成;消除反应;重排反应;氧化还原反应。 常常有些反应是几种反应类型的综合: (CH3)3CCH2Br+H2O→(CH3)3CCH2OH+(CH3)2C=CHCH3+(CH3)2COHCH2CH3 例1: SN E 重排
3CCH2Br+H2O→(CH3)3CCH2OH+(CH3)2C=CHCH3+(CH3)2COHCH2CH3. 例1: SN E 重排.")
2
反应机理(反应历程、反应机制)定义 对反应的整个过程(即由反应物到产物所经历的途径)的详细描述。 反应分几步进行(有几个基元反应),
哪一步是决定整个反应速率的反应定速步骤, 反应经过什么反应中间体, 反应条件在其中起什么作用等。 反应机理是根据很多实验事实总结后提出的,它有一定的适用范围,能解释很多实验事实,并能预测反应的发生。反应机理已成为有机结构理论的一部分。 在表述反应机理时,必须指出电子的流向,并规定用箭头表示一对电子的转移,用鱼钩箭头表示单电子的转移。

3
哈蒙德假说——能量最低原理 大多数过渡态与中间体能量最接近,若中间体相对稳定,则活化能低,反应活性大。即化学反应优先选择活化能最低、中间体相对稳定的途径进行。 如2-甲基丙烯加氯化氢的产物是2-甲基-2-氯丙烷,而不是2-甲基-1-氯丙烷。 叔C+稳定性 > 伯C+,意味着叔C+易形成,根据哈蒙德原理,相应的过渡态也易形成,所需能垒低,经叔碳正离子历程的反应产物也要比经由伯碳正离子的多,反应活性大。

4
较稳定,先形成,易反应。 + C H a.活性中间体:两个过渡态之间的自由基。 b.活化能Ea:过渡态与起始态的能量差。
3 2 + a.活性中间体:两个过渡态之间的自由基。 b.活化能Ea:过渡态与起始态的能量差。 c.决速步骤:生成烷基自由基的一步的慢步骤(所需活化能越高,反应速率越慢)是速率决定步骤。
是速率决定步骤。")
5
(一)反应类型总结
反应类型总结")
6
(二)主要反应类型的机理描述 1.自由基反应 (1)烷烃的自由基取代反应,其机理描述:
主要反应类型的机理描述 1.自由基反应 (1)烷烃的自由基取代反应,其机理描述:")
7
a.反应需要光照、高温或过氧化物等产生自由基的条件。 b.此类反应常经历三个过程。
此类反应的特点是: a.反应需要光照、高温或过氧化物等产生自由基的条件。 b.此类反应常经历三个过程。 c. 反应的活性中间体为自由基,产物种类与自由基种类和稳定性有很大关系。对于烃类化合物来说,其中不同类型的氢反应活性顺序为: 烯丙基氢或苄基氢>叔氢>仲氢>伯氢 自由基的稳定性:烯丙基或苄基>叔碳>仲碳>伯碳 使用NBS,可在温和的条件下进行α –卤代反应。 环烷烃的取代反应——自由基机理

8
(2)烯烃加溴化氢时过氧化物效应(自由基加成历程)
H-Cl、H-I与不对称烯烃加成无过氧化物效应的原因: H-Cl的离解能大(431KJ/mol),产生自由基比较困难。 H-I的离解能较小(297KJ/mol),较易产生I· ,但I·的活泼性差,难与烯烃迅速加成,却易自身结合成I2分子。 所以不对称烯烃与H-Cl和H-I加成时没有过氧化物效应。得到的加成产物仍服从马氏规则。
,产生自由基比较困难。 H-I的离解能较小(297KJ/mol),较易产生I· ,但I·的活泼性差,难与烯烃迅速加成,却易自身结合成I2分子。 所以不对称烯烃与H-Cl和H-I加成时没有过氧化物效应。得到的加成产物仍服从马氏规则。")
9
自由基加成历程: 链引发 由于自由基的稳定性为:3°R· > 2°R· > 1°R· > CH3· ,故其自由基加成的产物是反马氏规则的。

10
例1:依据下列反应事实,写出其可能的反应机理。
解:

11
2. 亲电反应 亲电类型的反应属于离子型反应,是亲电试剂进攻底物分子中带负电荷或电子云密度较高的原子(反应中心)而发生的反应。(一般经过碳正离子中间体) (1)亲电加成反应 这是烯烃、炔烃发生的典型的化学反应,机理: G为给电子基团(常为R基)。 反应试剂:X2(X+)、HX(H+)、H2O( H+)及其它无机酸。
。 反应试剂:X2(X+)、HX(H+)、H2O( H+)及其它无机酸。")
12
①Markevnikev规则 不对称的亲电试剂(即A不等于B)与不对称烯烃发生亲电加成反应时遵守Markevnikev规则。马氏规则可有两种表述方式: 1)在烯烃类化合物的亲电加成反应中,质子首先加在含氢较多的双键C原子上; 2)在烯烃类化合物的亲电加成反应中,亲电基团首先加在电子云密度比较高的双键C原子上; 第一种是经典的表述,其主要针对烯烃类化合物和可解离出质子的亲电试剂之间的反应,如:
在烯烃类化合物的亲电加成反应中,亲电基团首先加在电子云密度比较高的双键C原子上; 第一种是经典的表述,其主要针对烯烃类化合物和可解离出质子的亲电试剂之间的反应,如:")
13
但是,对于烯烃的其它衍生物,特别是烯键上带吸电基团的衍生物发生的亲电加成反应,马氏规则的经典表述就难以发挥作用。如:
这并不是违反马氏规则,而是遵守了马氏规则的本质表述。 三、四元环烷烃的加成反应——亲电加成反机理

14
②碳正离子中间体历程和碳正离子重排 Y- + 碳正离子机理进行的过程可表述如下:试剂首先解离成离子,正离子与烯烃反应形成碳正离子,这是决定反应速率的一步,π键断裂后,C—C键可以自由旋转,然后与带负电荷的离子结合,这时结合有两种可能,即生成顺式加成与反式加成两种产物。

15
碳正离子有可能重排为较稳定的碳正离子, 由于SN1和E1也是碳正离子中间体,也会生成取代或消除产物。

16
(I) 稳定性:叔C+﹥仲C+ (II) 1,2-甲基迁移、1,2-负氢迁移。重排为更稳定的碳正离子。 (40%) (60%) HCl
(40%) (60%) 0℃ HCl Cl- (I) Cl- (II) 氢迁移 稳定性:叔C+﹥仲C+
 (60%) 0℃ HCl. Cl- (I) Cl- (II) 氢迁移. 稳定性:叔C+﹥仲C+")
17
例2. 用反应机理解释所得的产物。 解:

18
例3. 用反应机理解释所得的产物。 解:

19
③鎓离子历程和反式加成产物 例4. 解: π-络合物

20
(2)亲电取代反应 亲电取代反应是芳环所发生的典型的化学反应。 ①机理: 加成—消除反应历程 E﹢= 、NO2 、SO3 、R 、 ﹢ X
亲电取代反应是芳环所发生的典型的化学反应。 芳环上的氢被亲电试剂取代的反应称为芳香亲电取代反应 ①机理: 加成—消除反应历程 E﹢= 、NO2 、SO3 、R 、 ﹢ X = O R—C 1)反应的第一步与亲电加成相似,活性中间体是苯环的芳香结构遭到破坏的环上带正电荷的离子: 2)与烯烃的加成反应不同的是,反应的第二步则是苯环因失去质子而变回到芳香结构。即结果亲电取代反应;
反应的第一步与亲电加成相似,活性中间体是苯环的芳香结构遭到破坏的环上带正电荷的离子: 2)与烯烃的加成反应不同的是,反应的第二步则是苯环因失去质子而变回到芳香结构。即结果亲电取代反应;")
21
3)除了磺化反应外,亲电试剂均是在其它试剂(催化剂)的帮助下产生的,且催化剂都是酸。
Br2 + FeBr3 Br+ + FeBr4 -

22
②傅-克烷基化反应的重排和可逆 (66%) 亲电试剂异丁基正离子重排为更稳定的叔丁基正离子,
烷基化反应为可逆反应,故烷基苯可进行歧化反应,即一分子烷基苯脱烷基变成苯,另一分子烷基苯增加烷基变成二烷基苯。如:

23
例5. 解: 思考题:写出下列反应的反应历程:

24
③定位效应 当芳环上原先有取代基时,会对新进入的取代基的取代位置产生影响,此即定位基(原有取代基称做定位基)的定位效应;定位基不仅影响新亲电基团的进入位置,也对反应的活性产生影响。 基团在苯环上的定位效应及对反应活性的影响分作三类。 1)活化的邻对位定位基: -O-,-NH2,-OH,-OCH3,-X,-R,等。 致活的原因是通过(诱导或共轭)给电,导致苯环的电子云密度提高,更有利于亲电基团的进攻。或给电子基团使碳正离子中间体稳定,反应活性增大。
活化的邻对位定位基: -O-,-NH2,-OH,-OCH3,-X,-R,等。 致活的原因是通过(诱导或共轭)给电,导致苯环的电子云密度提高,更有利于亲电基团的进攻。或给电子基团使碳正离子中间体稳定,反应活性增大。")
25
例:甲基是致活基!

26
为什么新引入基上第一类定位基的o-、p-? 分析中间体的稳定性
假设硝酰基换成了给电子基,如甲基,可看到E进攻致活定位基的邻位和对位时,基团的给电子达到最大效应,碳正离子中间体较稳定。 δ+ δ+ δ+ δ+ δ+ δ+ δ+ δ+ δ+ 为什么新引入基上第二类定位基的m-? 当E+进攻硝基的不同位置时,邻、对位取代中间体中都有较正部分直接与吸电子基相连而不稳定。比较起来只有间位较有利。

27
2) 钝化的间位定位基,如:-NO2、-SO3H、-COOH、-CO-、-CHO、-CN等;这些基团使亲电基团进入自身的间位,且使其所连的苯环反应的活性降低;降低活性的原因是通过吸电的共轭效应使得苯环的电子云密度降低, 3)弱钝化的邻对位定位基,苯环上连有卤素时即发挥此种作用;卤素为邻对位定位基的原因是p-π 共轭效应导致卤素邻对位C上的电子云密度高于间位;而卤素钝化的原因是吸电的诱导效应使苯环的电子云密度降低。 –I < +C -OH -OR -NH2 -NO2、 -SO3H、 -COOH等、 -X ﹥ ﹥ 苯 –I > +C ﹥ –I ,—C 邻、 对 位 定 位 间位 定 位
弱钝化的邻对位定位基,苯环上连有卤素时即发挥此种作用;卤素为邻对位定位基的原因是p-π 共轭效应导致卤素邻对位C上的电子云密度高于间位;而卤素钝化的原因是吸电的诱导效应使苯环的电子云密度降低。 –I < +C. -OH. -OR. -NH2. -NO2、 -SO3H、 -COOH等、 -X. ﹥ ﹥ 苯. –I > +C. ﹥ –I ,—C. 邻、 对 位 定 位. 间位 定 位.")
28
4)空间效应 第三取代基一般不进入1,3-二取代苯的2位——空阻!
空间效应 第三取代基一般不进入1,3-二取代苯的2位——空阻!")
29
(3) 赫尔-乌尔哈-泽林斯基反应 在三氯化磷或三溴化磷等催化剂的作用下,卤素取代羧酸α-H的反应称为 赫尔-乌尔哈-泽林斯基反应
(3) 赫尔-乌尔哈-泽林斯基反应 在三氯化磷或三溴化磷等催化剂的作用下,卤素取代羧酸α-H的反应称为 赫尔-乌尔哈-泽林斯基反应 PBr3 -HBr RCHCOOH RCH2COOH + Br2 Br 羧酸α—H被Br+取代 亲电试剂
 赫尔-乌尔哈-泽林斯基反应. 在三氯化磷或三溴化磷等催化剂的作用下,卤素取代羧酸α-H的反应称为 赫尔-乌尔哈-泽林斯基反应. PBr3. -HBr. RCHCOOH. RCH2COOH + Br2. Br. 羧酸α—H被Br+取代. 亲电试剂.")
30
3. 亲核类型的反应 (1)亲核取代反应 ①单分子亲核取代机理(SN1)
有机化学中的亲核反应属于离子反应类型,即带负电荷或部分负电荷的亲核基团(试剂或其它反应物)进攻底物中带正电荷或部分正电荷的原子(反应中心)而发生的反应。 (1)亲核取代反应 发生亲核取代反应的有卤代烃(碱性条件)、醇(酸性条件)等。 ①单分子亲核取代机理(SN1) 其机理可以表示为:
进攻底物中带正电荷或部分正电荷的原子(反应中心)而发生的反应。 (1)亲核取代反应. 发生亲核取代反应的有卤代烃(碱性条件)、醇(酸性条件)等。 ①单分子亲核取代机理(SN1) 其机理可以表示为:")
31
1) SN1的立体化学特点: 产物是外消旋体。 构型保持 中间体 构型转化
 SN1的立体化学特点: 产物是外消旋体。 构型保持 中间体 构型转化")
32
构型保持和构型翻转 构型翻转 构型保持 (R)-2-溴辛烷 []D= - 34.6o (S)-2-辛醇 []D= + 9.9o
HO- (R)-2-溴辛烷 []D= o (S)-2-辛醇 []D= + 9.9o (R)-2-辛醇 []D= - 9.9o 构型翻转 构型保持 若反应涉及到一个不对称碳原子上的一根键的变化,则将新键在旧键断裂方向形成的情况称为构型保持,而将新键在旧键断裂的相反方向形成的情况称为构型翻转。这种构型的翻转也称为Walden转换。
![构型保持和构型翻转 构型翻转 构型保持 (R)-2-溴辛烷 []D= o (S)-2-辛醇 []D= + 9.9o](http://slidesplayer.com/slide/11130435/59/images/32/%E6%9E%84%E5%9E%8B%E4%BF%9D%E6%8C%81%E5%92%8C%E6%9E%84%E5%9E%8B%E7%BF%BB%E8%BD%AC+%E6%9E%84%E5%9E%8B%E7%BF%BB%E8%BD%AC+%E6%9E%84%E5%9E%8B%E4%BF%9D%E6%8C%81+%EF%BC%88R%EF%BC%89-2-%E6%BA%B4%E8%BE%9B%E7%83%B7+%5B%EF%81%A1%5DD%3D+o+%EF%BC%88S%EF%BC%89-2-%E8%BE%9B%E9%86%87+%5B%EF%81%A1%5DD%3D+%2B+9.9o.jpg "HO- (R)-2-溴辛烷. []D= o. (S)-2-辛醇. []D= + 9.9o. (R)-2-辛醇. []D= - 9.9o. 构型翻转. 构型保持. 若反应涉及到一个不对称碳原子上的一根键的变化,则将新键在旧键断裂方向形成的情况称为构型保持,而将新键在旧键断裂的相反方向形成的情况称为构型翻转。这种构型的翻转也称为Walden转换。")
33
2) 影响反应的因素:碳正离子结构的稳定, 3) SN1机理常伴有重排反应: 因此SN1反应的活性顺序:
苄基型,烯丙基型>叔卤代烷 > 仲卤代烷 > 伯卤代烷 > CH3X 3) SN1机理常伴有重排反应:
 SN1机理常伴有重排反应:")
34
②双分子亲核取代机理(SN2) 过渡态 SN2反应机理的主要特点为:
1)构型翻转:在过渡态中,离去基团并未从中心C上脱下来,因此亲核基团只能从离去基团的背面进攻;这样,SN2反应的立体化学特点为构型转化。 2)空间效应 速度决定步骤是过渡态。在这个步骤中需要底物和亲核试剂同时参加,拥挤,所以SN2反应的活性: CH3X > 伯卤代烷 > 仲卤代烷 > 叔卤代烷 3)SN2反应不发生重排
构型翻转:在过渡态中,离去基团并未从中心C上脱下来,因此亲核基团只能从离去基团的背面进攻;这样,SN2反应的立体化学特点为构型转化。 2)空间效应 速度决定步骤是过渡态。在这个步骤中需要底物和亲核试剂同时参加,拥挤,所以SN2反应的活性: CH3X > 伯卤代烷 > 仲卤代烷 > 叔卤代烷. 3)SN2反应不发生重排.")
35
氯乙烯型、氯苯分子中氯原子的-I效应和p,π-共轭效应共同影响的结果,导致C―Cl键键长缩短,键的离解能增大。所以,卤苯型化合物的卤原子表现出较低的反应活性,例如:在一般条件下,卤原子不易被亲核试剂取代;与AgNO3-alc. 溶液不反应;在F-C反应中,不能象RX那样作为烃基化试剂使用。 烯丙型、苄基型卤代物,其X原子比较活泼。它们无论进行SN1反应还是SN2反应都比较容易,尤其是SN1反应。 C6H5-CH2+ X 不易离去SN反应难 X 易离去,SN反应活性大

36
p-共轭正电荷分散稳定 p-共轭C―Cl键键长缩短, 键的离解能增大 氯苯 + + + + + + 烯丙基正碳离子
苄基正碳离子 p-共轭正电荷分散稳定 p-共轭C―Cl键键长缩短, 键的离解能增大 氯苯

37
卤代烃的取代反应一览 水解反应 醇解反应 氰解反应 增加1个C 氨解反应

38
生成新的 C-C 键:

39
1,2-环氧化合物的开环反应 环氧乙烷类化合物的三元环分子中存在张力,极易与多种试剂反应,把环打开。 酸催化开环反应时,首先环氧化物的氧原子质子化,然后亲核试剂向C−O键的碳原子的背后进攻取代基较多的环碳原子,具有SN1的性质,电子效应控制了产物,空间因素不重要。 碱性开环时,亲核试剂选择进攻取代基较少的环碳原子,C−O键的断裂与亲核试剂和环碳原子之间键的形成几乎同时进行,这是一个SN2反应,空间效应控制了反应。

40
1, 2- 环氧化合物在酸性条件下开环反应的反应机理
H+ 18 -H+ + + 18

41
1,2 - 环氧化合物碱性开环反应的反应机理

42
③芳环上的亲核取代反应 芳环上的一个基团被一个亲核试剂取代,称为芳环上的亲核取代反应。 + NaCN C6H5CN + NaCl

43
1)加成-离去机理 60℃ 200℃ 高压 100℃ 130℃
加成-离去机理 60℃ 200℃ 高压 100℃ 130℃")
44
其它吸电子基如:-SO3H、-CN、-+NR3、-COR、-COOH、-CHO等也有类似影响
实例: -X HX -NO2 HNO2

45
选择取代: 在卤原子的邻,对位上强第二类定位基越多,亲核取代越容易。 卤原子活性:F > Cl > Br > I

46
卤原子活性:F > Cl > Br > I ,电负性越大越有利于负电荷的分散。
加成―离去机理 和苯环的亲电取代反应相似,不同的是第一步亲核加成,形成碳负离子中间体;第二步卤原子离去。由于中间体是带负电荷的离子,中心碳的邻、对位有吸电子基反而促进反应进行。 慢 卤原子活性:F > Cl > Br > I ,电负性越大越有利于负电荷的分散。

47
2) 消去―加成机理(苯炔机理) 特点:新引入的基团不仅进入卤原子原来的位置,还进入了 卤原子的邻位。 苯炔活性中间体 该历程的前两步是从氯苯中消去一分子HCl,生成苯炔中间体,然后在苯炔的碳碳三键上加进了一分子的NH3,故称消去―加成机理;又因该类反应是经过苯炔中间体完成的,故又称苯炔机理。

48
2. 用KN(C2H5)2 - (C2H5)2NH处理N-甲基-N-(2’-邻氯苯
练习:1.完成下列反应: 2. 用KN(C2H5)2 - (C2H5)2NH处理N-甲基-N-(2’-邻氯苯 基乙基)胺和N-甲基-N-(2’-间氯苯基乙基)胺得到高产率的 同一产物,其分子式为C9H11N。试回答:(1) 这个产物是 什么?(2) 写出反应历程。
2 - (C2H5)2NH处理N-甲基-N-(2’-邻氯苯. 基乙基)胺和N-甲基-N-(2’-间氯苯基乙基)胺得到高产率的. 同一产物,其分子式为C9H11N。试回答:(1) 这个产物是. 什么?(2) 写出反应历程。")
49
完成反应式

50
(2)亲核加成反应 反应物 过渡态 反应中间体 产物 平面三角形 四面体 四面体 影响反应活性大小的因素:
是醛、酮类化合物典型的反应形式,其机理为: 反应物 过渡态 反应中间体 产物 平面三角形 四面体 四面体 影响反应活性大小的因素: ①羰基C是反应的活性中心。凡能增加羰基C正电荷的因素,均使反应速度增加,凡是降低羰基C正电荷的因素则使反应速度降低。简言之:拉电子有利,推电子不利。 ②亲核加成反应的快慢还与羰基C所连基团的大小有关,体积越大,速度越慢。如醛比酮活性大。

51
亲核加成小结 合成 缩醛,环状半缩醛,保护羰基 鉴定,分离提纯 鉴定,提纯,确定结构 醛醇的缩合: 羰基与胺衍生物缩合:

52
例6. 写出下面反应的反应历程。 解:

53
岐化反应 交叉歧化反应: 在交叉歧化反应中,总是甲醛被氧化,其它醛被还原.

54
反应历程: 酸 碱

55
(3)亲核加成-离去 (羧酸及其衍生物的亲核取代) 亲核加成-消除机理 + + 反应活性:酰卤>酸酐>酯>酰胺
亲核加成-离去 (羧酸及其衍生物的亲核取代) 亲核加成-消除机理 + + 反应活性:酰卤>酸酐>酯>酰胺")
56
酯的碱性水解(又称皂化反应) 反应机理 快 慢 四面体中间体 是负离子
 反应机理 快 慢 四面体中间体 是负离子")
57
(4)碳负离子对羰基的亲核加成反应或加成-消除反应
亲核试剂:羰基化合物α-氢的电离后形成的碳负离子 ①羟醛缩合反应(实质上是羰基的亲核加成): 1)反应条件:稀碱,至少含有α氢的醛存在; 2)产物:β-羟基醛(酮)或αβ-不饱和醛(酮)。
: 1)反应条件:稀碱,至少含有α氢的醛存在; 2)产物:β-羟基醛(酮)或αβ-不饱和醛(酮)。")
59
例7. 写出下列反应的反应历程 解:

61
② Reformatsky 反应(羰基的亲核加成反应)
α- 卤代酸酯与醛或酮在乙醚、苯、甲苯等惰性溶剂中与锌粉反应,产物水解后得到β- 醇酸酯。 意义:有机锌试剂只与醛酮的羰基反应,保留了酯羰基。不能用镁代替锌,因有机镁试剂太活泼,与酯也发生反应。 机理:

62
③ Claisen酯缩合反应(酯的亲核加成-离去机理)
酯分子中的α-活泼氢在醇钠的催化作用下,可与另一分子酯脱去一分子醇而互相缩合,这类反应称为Claisen Condensation反应。

63
机理:亲核加成-离去——亲核取代 亲核试剂生成 亲核加成 离去 结果

64
两种不同的有α-H的酯的缩合产物复杂,无实用价值。 无α-H的酯与有α-H的酯的缩合产物纯,有合成价值。
分子之间缩合称为克莱森(Claisen)酯缩合反应。 分子内的酯缩合叫Dieckmann condensation。 两种不同的有α-H的酯的缩合产物复杂,无实用价值。 无α-H的酯与有α-H的酯的缩合产物纯,有合成价值。 反应特点: 底物:含有α–氢的酯作为Nu 强碱: 醇钠 产物: 生成β–酮酸酯 增长碳链的反应
酯缩合反应。 分子内的酯缩合叫Dieckmann condensation。 两种不同的有α-H的酯的缩合产物复杂,无实用价值。 无α-H的酯与有α-H的酯的缩合产物纯,有合成价值。 反应特点: 底物:含有α–氢的酯作为Nu. 强碱: 醇钠. 产物: 生成β–酮酸酯. 增长碳链的反应.")
65
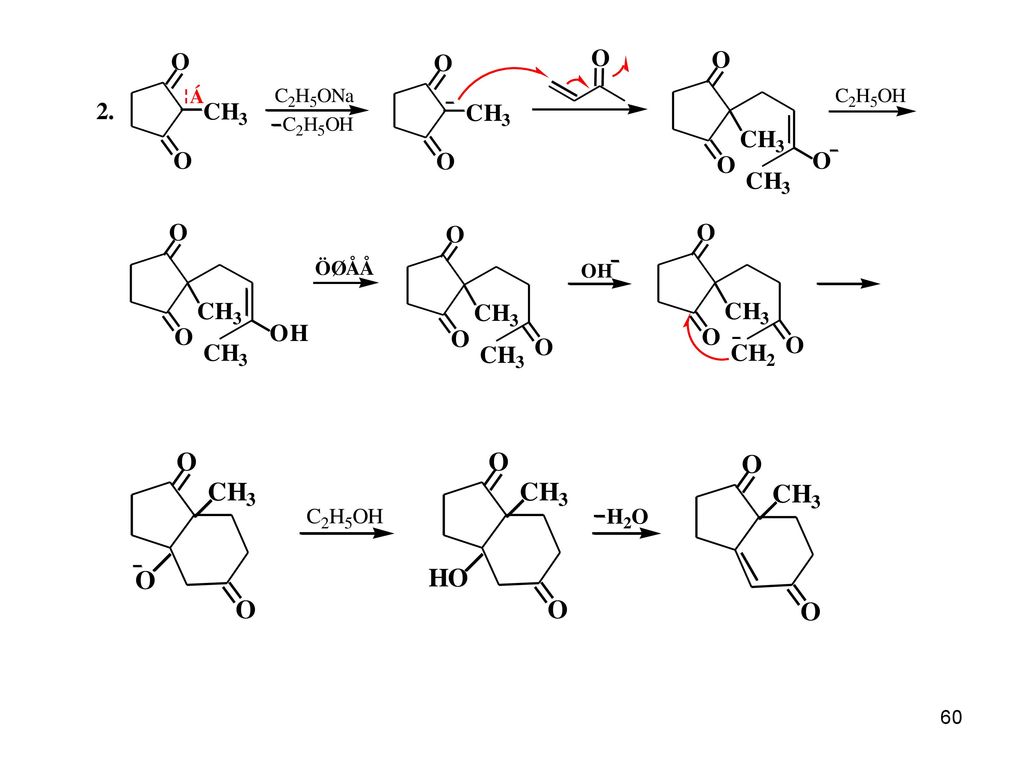
④.迈克尔(Michael)加成反应 碳-碳双键上有吸电子基如羰基时,其亲电性减弱而亲核性加强,能够接受亲核试剂——碳负离子的进攻。即,碳负离子与活性双键的加成反应,称为迈克尔反应。 RCOCH2-CH2CH2COR 合成1,5-二羰基化合物 Β-二酮、苯乙腈、乙酰乙酸乙酯和丙二酸二乙酯等在碱性催化剂的存在下都能与双键起加成反应:

67
反应机理: ——1,4-加成 例:

68
芳醛和脂肪族酸酐在碱金属盐共热下的缩合反应
⑤. Perkin 反应 芳醛和脂肪族酸酐在碱金属盐共热下的缩合反应 -H2O

69
4. 消除反应 RX、ROH、RNH2在一定条件下可发生E反应。

70
(1)E1机理 ①以Saytzeff产物为主 查依耶夫定则:生成热力学稳定的烯烃为主,即形成双键上有烃基最多的烯烃(超共轭来解释。
E1机理 ①以Saytzeff产物为主 查依耶夫定则:生成热力学稳定的烯烃为主,即形成双键上有烃基最多的烯烃(超共轭来解释。")
71
②E1和SN1的第一步完全一样,碳正离子也有重排的可能,如醇在酸性条件下生成R+,重排现象频繁。

72
例8:写出下面反应的反应历程。 解:

73
思考题:

74
②无论是发生E1反应还是发生E2反应,RX的反应活性均为:3oRX 2oRX 1oRX
Saytzeff烯 ①过渡态已有烯烃的雏形,所以产物以Saytzeff烯为主 ②无论是发生E1反应还是发生E2反应,RX的反应活性均为:3oRX 2oRX 1oRX E1反应,3oC+最易形成。 E2反应, 3oRX提供较多的-H。

75
③消除的立体化学:反式共平面消除(从过渡态可加以理解)
例如: H 多 少 H 不论什么机理有可能成共轭烯烃一般成共轭烯烃。

76
(3)季铵碱的热消除——Hoffmann烯
反应机理: E2反应 当有两种以上b-H时, 生成双键上烃基少的烯烃——Hoffmann烯,原因:空间位阻

77
但若β-C上有芳基、乙烯基等时, 则反应不按Hoffmann规律进行。
此反应常用于测定胺的结构。如:





 3、一些常用的不饱和基团(烯基)>")

 一元卤代烃 卤代烃 不饱和卤代烃 二元卤代烃 卤代烃 卤代芳烃>")





![第六章 卤代烃 亲核取代反应(4学时) [目的要求]: 1. 掌握卤代烃的命名, 了解卤代烃的分类。 2. 掌握卤代烃的性质和制备;](/61/11444981/big_thumb.jpg "第六章 卤代烃 亲核取代反应(4学时) [目的要求]: 1. 掌握卤代烃的命名, 了解卤代烃的分类。 2. 掌握卤代烃的性质和制备;>")

重要有机反应的反应机理.>")

基化反应 醛(酮)及酯的缩合反应 生成碳碳双键的反应 成环的反应 其他类型的负碳离子反应>")




 朱传方 2007.11..>")
