Download presentation
Presentation is loading. Please wait.

1
Review: 开环在药物合成的主要应用特征
ii) 在一个双环或多环分子中,断裂被两个环所共用的化学键,可形成中等或大环的分子,而这些中环和大环分子则很难用其他方法来制备。 用途独特! i) 在开环反应产物中,被断裂的化学键每一端原子上都带有官能团;用于合成双官能团分子,其双官能团被几个其他的原子隔开。 进一步用于合成杂环。
 在一个双环或多环分子中,断裂被两个环所共用的化学键,可形成中等或大环的分子,而这些中环和大环分子则很难用其他方法来制备。 用途独特! i) 在开环反应产物中,被断裂的化学键每一端原子上都带有官能团;用于合成双官能团分子,其双官能团被几个其他的原子隔开。 进一步用于合成杂环。")
2
Chapter 6. Reduction and oxidation 2015,11,15
第六章 还原和氧化 Chapter 6. Reduction and oxidation 2015,11,15

3
还原反应 1 催化氢化反应 2 金属氢化物还原 3 电子转移反应 4 特殊官能团的还原 5 碳-杂原子键的还原断裂 6 环氧化物的还原开环
1 催化氢化反应 2 金属氢化物还原 3 电子转移反应 4 特殊官能团的还原 5 碳-杂原子键的还原断裂 6 环氧化物的还原开环 7 α,β-不饱和羰基化合物的还原 8 共轭二烯的还原 9 芳香和杂芳香化合物的还原

4
1 催化氢化反应 1.1 非均相催化反应 温度和压力:1-4 atom
1 催化氢化反应 1.1 非均相催化反应 温度和压力:1-4 atom 催化剂:雷尼镍、铂[通常是通过使用PtO2-亚当(Adams)催化剂在催化氢化时就地而产生]、或是附着在载体上的钯或铑,载体是用来降低催化剂活性,可是碳、硫酸钡或碳酸钙。
催化剂在催化氢化时就地而产生]、或是附着在载体上的钯或铑,载体是用来降低催化剂活性,可是碳、硫酸钡或碳酸钙。")
5
•表8.1 催化氢化的产物 官能团 氢化产物 RCOCl RCHO RNO2 RNH2 RC≡CR RCH=CHR(Z) RCHOR
•表8.1 催化氢化的产物 官能团 氢化产物 RCOCl RCHO RNO2 RNH2 RC≡CR RCH=CHR(Z) RCHOR CH2OH RCH=CHR RCH2CH2R RCOR RCH(OH)R ArCH2X ArCH3 RC≡N RCH2NH2 RCOOR’ RCH2OH + R’OH RCONHR RCH2NHR
 RCHOR. CH2OH. RCH=CHR. RCH2CH2R. RCOR. RCH(OH)R. ArCH2X. ArCH3. RC≡N. RCH2NH2. RCOOR’ RCH2OH + R’OH. RCONHR. RCH2NHR.")
6
非均相催化反应的特点 溶剂也可以影响到催化剂的活性: 从中性的非极性溶剂如环己烷到极性的酸性溶剂如乙酸,催化剂的活性将会依次递增。
非均相催化剂氢化可导致底物异构化。

7
Examples(old) 罗格列酮 (Rosiglitazone)直线式合成最后一步
吡格列酮 (Pioglitazone) 直线式合成最后一步
 直线式合成最后一步.")
8
Example (old) 非典型安定药利培酮(Risperidone)中间体的合成
6,7,8,9-tetrahydro-3-(2-hydroxyethyl)- 2-methylpyrido[1,2-a]pyrimidin-4-one Janssen: Risperdal® (1993)
- 2-methylpyrido[1,2-a]pyrimidin-4-one. Janssen: Risperdal® (1993)")
9
Example (old) 美容药:非那甾胺 (Finasteride)中间体的合成
(4aR,4bS,6aS,7S,9aS,9bS,11aR)-hexadecahydro-4a,6a-dimethyl-2-oxo-1H-indeno[5,4-f]quinoline-7-carboxylic acid Merk: Propecia® (1997)
-hexadecahydro-4a,6a-dimethyl-2-oxo-1H-indeno[5,4-f]quinoline-7-carboxylic acid. Merk: Propecia® (1997)")
10
1.2 均相催化反应 利用均相催化剂可使异构化程度降低至最少,例如使用三(三苯基膦基)-氯化铑。
1.2 均相催化反应 利用均相催化剂可使异构化程度降低至最少,例如使用三(三苯基膦基)-氯化铑。 因为和在非均相反应中的对应物相比,中间体络合物不大易容易发生重排。
-氯化铑。 因为和在非均相反应中的对应物相比,中间体络合物不大易容易发生重排。")
11
均相催化反应的特点 均相催化剂不容易从反应混和物中被分离, 可用聚合物-键合的类似物兼有容易分离除去和形成产物具有高纯度的双重特点:

12
1.3 转移催化氢化反应 在这种方法中,氢的来源不是氢元素本身,而是一种在催化剂的作用下可能发生的脱氢化作用的化合物。因此,氢原子从供体转移给催化剂,然后再传递给要被还原的底物。

13
氢供体的种类 氢的供体可以是有机化合物(如:环己烯、2-丙醇或甲酸)或者无机化合物(如:肼或硼氢化钠),而催化剂可以是非均相或是均相的。
比较更传统的技术而言,该方法明显的优点就是不使用气态氢,避免附带危险。

14
2 金属氢化物还原反应 某些金属氢化物是氢负离子(H-)合成子的合成等价物,因而是优先和缺电子中心发生反应的强效还原性试剂。然而,碱性更强的氢化物(例如:NaH和CaH2)却不是还原剂。 在市场上容易买到的多种氢化物还原剂(见表8.2)中,有一些与水发生剧烈反应、与醇作用很容易发生反应,因此,该类反应必须在无水醚类或烃类溶剂中操作进行。对于每一种试剂所使用的最常见的溶剂也列在表8.2中 。
中,有一些与水发生剧烈反应、与醇作用很容易发生反应,因此,该类反应必须在无水醚类或烃类溶剂中操作进行。对于每一种试剂所使用的最常见的溶剂也列在表8.2中 。")
15
表2 用于金属氢化物还原的溶剂 编号 金属氢化物 溶剂 1 LiAlH4 乙醚,四氢呋喃,二甘醇二甲醚 2 LiAlH[OC(CH3)3]3
表2 用于金属氢化物还原的溶剂 编号 金属氢化物 溶剂 1 LiAlH4 乙醚,四氢呋喃,二甘醇二甲醚 2 LiAlH[OC(CH3)3]3 四氢呋喃,二甘醇二甲醚 3 NaAlH2(OCH2CH2OCH3)2[RED-Al] 苯,甲苯,二甲苯 4 NaBH4 水,乙醇,二甘醇二甲醚 5 NaBH3(CN) 水,甲醇,二甲基亚砜 6 LiBH4 7 AlH3 乙醚,四氢呋喃, 8 AlH[(CH2CH(CH3)2]2[DIBAL-H] 甲苯,CH3O(CH2)2OCH3
![表2 用于金属氢化物还原的溶剂 编号 金属氢化物 溶剂 1 LiAlH4 乙醚,四氢呋喃,二甘醇二甲醚 2 LiAlH[OC(CH3)3]3](http://slidesplayer.com/slide/11130619/59/images/15/%E8%A1%A82+%E7%94%A8%E4%BA%8E%E9%87%91%E5%B1%9E%E6%B0%A2%E5%8C%96%E7%89%A9%E8%BF%98%E5%8E%9F%E7%9A%84%E6%BA%B6%E5%89%82+%E7%BC%96%E5%8F%B7+%E9%87%91%E5%B1%9E%E6%B0%A2%E5%8C%96%E7%89%A9+%E6%BA%B6%E5%89%82+1+LiAlH4+%E4%B9%99%E9%86%9A%EF%BC%8C%E5%9B%9B%E6%B0%A2%E5%91%8B%E5%96%83%EF%BC%8C%E4%BA%8C%E7%94%98%E9%86%87%E4%BA%8C%E7%94%B2%E9%86%9A+2+LiAlH%5BOC%28CH3%293%5D3.jpg "表2 用于金属氢化物还原的溶剂. 编号. 金属氢化物. 溶剂. 1. LiAlH4. 乙醚,四氢呋喃,二甘醇二甲醚. 2. LiAlH[OC(CH3)3]3. 四氢呋喃,二甘醇二甲醚. 3. NaAlH2(OCH2CH2OCH3)2[RED-Al] 苯,甲苯,二甲苯. 4. NaBH4. 水,乙醇,二甘醇二甲醚. 5. NaBH3(CN) 水,甲醇,二甲基亚砜. 6. LiBH4. 7. AlH3. 乙醚,四氢呋喃, 8. AlH[(CH2CH(CH3)2]2[DIBAL-H] 甲苯,CH3O(CH2)2OCH3.")
16
NaAlH2(OCH2CH2OCH3)2[RED-Al] AlH[(CH2CH(CH3)2]2[DIBAL-H]
表3 金属氢化物还原的产物 还原反应 还 原 剂 LiAlH4 LiAlH[OC(CH3)3]3 NaAlH2(OCH2CH2OCH3)2[RED-Al] NaBH4 NaBH3(CN) LiBH4 AlH3 AlH[(CH2CH(CH3)2]2[DIBAL-H] RCHO → RCH2OH √ RCOR → RCH(OH)RR COCl → RCH2OH (a) 醛 内酯 → 二醇 × (b) 慢 (c) 乳醇 环氧化物 →醇 RCO2R’→RCH2OH + (d) 苯基酯得醛 R’OHRCOOH→RCH2OH RCONR2→RCH2NR2 (e)一些酰胺成醛 RC≡N→RCH2NH2 RNO2→RNH2 R=Aliphatic R=Aromatic,则偶氮化 RX(g) → RH X=F, Cl, Br, I, OSO2R’ RC≡CR → RCH=CHR(Z) (a) 还原只进行到醛的阶段。 (b) 反应非常慢。 (c) 反应仅进行到乳醇(邻位羟基内醚 )的阶段。 (d) 苯基酯得到醛。 (e) 一些酰胺被还原成醛。 (f) R是脂肪族的,如果R是芳香族的,则形成偶氮芳烃。 (s) X =卤素或OSO2R’。
![NaAlH2(OCH2CH2OCH3)2[RED-Al] AlH[(CH2CH(CH3)2]2[DIBAL-H]](http://slidesplayer.com/slide/11130619/59/images/16/NaAlH2%28OCH2CH2OCH3%292%5BRED-Al%5D+AlH%5B%28CH2CH%28CH3%292%5D2%5BDIBAL-H%5D.jpg "表3 金属氢化物还原的产物. 还原反应. 还 原 剂. LiAlH4. LiAlH[OC(CH3)3]3. NaAlH2(OCH2CH2OCH3)2[RED-Al] NaBH4. NaBH3(CN) LiBH4. AlH3. AlH[(CH2CH(CH3)2]2[DIBAL-H] RCHO → RCH2OH. √ RCOR → RCH(OH)RR. COCl → RCH2OH. (a) 醛. 内酯 → 二醇. × (b) 慢. (c) 乳醇. 环氧化物 →醇. RCO2R’→RCH2OH + (d) 苯基酯得醛. R’OHRCOOH→RCH2OH. RCONR2→RCH2NR2. (e)一些酰胺成醛. RC≡N→RCH2NH2. RNO2→RNH2. R=Aliphatic. R=Aromatic,则偶氮化. RX(g) → RH. X=F, Cl, Br, I, OSO2R’ RC≡CR → RCH=CHR(Z) (a) 还原只进行到醛的阶段。 (b) 反应非常慢。 (c) 反应仅进行到乳醇(邻位羟基内醚 )的阶段。 (d) 苯基酯得到醛。 (e) 一些酰胺被还原成醛。 (f) R是脂肪族的,如果R是芳香族的,则形成偶氮芳烃。 (s) X =卤素或OSO2R’。")
17
Example鲁比前列酮中间体的合成 各种还原反应的选择性应用: 鲁比前列酮的合成路线三 p355
不饱和二氟代酮 ,在乙酸乙酯中用H2/Pd/C还原化合物其侧链的双键,得饱和酮 ,用NaBH4的甲醇溶液还原得仲醇 ,进一步用DIBAL的甲苯溶液还原得半缩醛 。 鲁比前列酮的合成路线三 p355

18
LiAlH4、RED-AL®和AlH3 LiAlH4、RED-AL®和AlH3是非选择性的试剂。只有在α, β-不饱和羰基化合物的情况下AlH3才优于LiAlH4。事实上,选择性更强的试剂DIBAL-H也许是一种更好的选择。 RED-AL®和 LiAlH4比较具有如下的优点:i) 在潮湿空气中不会着火,且在干燥的空气中能稳定存在,ii) 高达200℃时,该化合物具有热力学稳定性,iii) 很容易溶于芳香性烃类溶剂。氰基硼氢化钠则是一种具有高度选择性的还原试剂:如,它甚至可在醛的存在下用于还原一级卤代烃。
 在潮湿空气中不会着火,且在干燥的空气中能稳定存在,ii) 高达200℃时,该化合物具有热力学稳定性,iii) 很容易溶于芳香性烃类溶剂。氰基硼氢化钠则是一种具有高度选择性的还原试剂:如,它甚至可在醛的存在下用于还原一级卤代烃。")
19
溶剂影响反应选择 硼氢化钠在二甘醇二甲醚溶液中是一个非常温和的试剂,它能还原醛而不能还原酮。这种选择性也可通过使用三乙酸基硼氢化锂作为还原剂来实现。氯化铈(III)的作用是增加甲醇的酸性,结果则是促进具有更高选择性的物种形成,其结构式为[BHx(OMe)(4-x)]-。 为得到预期选择性,选择试剂、溶剂和反应条件极为重要。不断引入一些选择性的新结构的氢化物,包括那些可用于不对称合成的铝锂氢化物。
![溶剂影响反应选择 硼氢化钠在二甘醇二甲醚溶液中是一个非常温和的试剂,它能还原醛而不能还原酮。这种选择性也可通过使用三乙酸基硼氢化锂作为还原剂来实现。氯化铈(III)的作用是增加甲醇的酸性,结果则是促进具有更高选择性的物种形成,其结构式为[BHx(OMe)(4-x)]-。](http://slidesplayer.com/slide/11130619/59/images/19/%E6%BA%B6%E5%89%82%E5%BD%B1%E5%93%8D%E5%8F%8D%E5%BA%94%E9%80%89%E6%8B%A9+%E7%A1%BC%E6%B0%A2%E5%8C%96%E9%92%A0%E5%9C%A8%E4%BA%8C%E7%94%98%E9%86%87%E4%BA%8C%E7%94%B2%E9%86%9A%E6%BA%B6%E6%B6%B2%E4%B8%AD%E6%98%AF%E4%B8%80%E4%B8%AA%E9%9D%9E%E5%B8%B8%E6%B8%A9%E5%92%8C%E7%9A%84%E8%AF%95%E5%89%82%EF%BC%8C%E5%AE%83%E8%83%BD%E8%BF%98%E5%8E%9F%E9%86%9B%E8%80%8C%E4%B8%8D%E8%83%BD%E8%BF%98%E5%8E%9F%E9%85%AE%E3%80%82%E8%BF%99%E7%A7%8D%E9%80%89%E6%8B%A9%E6%80%A7%E4%B9%9F%E5%8F%AF%E9%80%9A%E8%BF%87%E4%BD%BF%E7%94%A8%E4%B8%89%E4%B9%99%E9%85%B8%E5%9F%BA%E7%A1%BC%E6%B0%A2%E5%8C%96%E9%94%82%E4%BD%9C%E4%B8%BA%E8%BF%98%E5%8E%9F%E5%89%82%E6%9D%A5%E5%AE%9E%E7%8E%B0%E3%80%82%E6%B0%AF%E5%8C%96%E9%93%88%28III%29%E7%9A%84%E4%BD%9C%E7%94%A8%E6%98%AF%E5%A2%9E%E5%8A%A0%E7%94%B2%E9%86%87%E7%9A%84%E9%85%B8%E6%80%A7%EF%BC%8C%E7%BB%93%E6%9E%9C%E5%88%99%E6%98%AF%E4%BF%83%E8%BF%9B%E5%85%B7%E6%9C%89%E6%9B%B4%E9%AB%98%E9%80%89%E6%8B%A9%E6%80%A7%E7%9A%84%E7%89%A9%E7%A7%8D%E5%BD%A2%E6%88%90%EF%BC%8C%E5%85%B6%E7%BB%93%E6%9E%84%E5%BC%8F%E4%B8%BA%5BBHx%28OMe%29%284-x%29%5D-%E3%80%82.jpg "为得到预期选择性,选择试剂、溶剂和反应条件极为重要。不断引入一些选择性的新结构的氢化物,包括那些可用于不对称合成的铝锂氢化物。")
20
Example 抗心绞痛药: 伊伐布雷定(Ivabradine)中间体合成
法国施维亚(Servier)公司:Procoralan® (2005) (Z)-3-(2-bromo-4,5-dimethoxyphenyl)-2-cyanoacrylic acid
公司:Procoralan® (2005) (Z)-3-(2-bromo-4,5-dimethoxyphenyl)-2-cyanoacrylic acid.")
21
Example 抗血小板聚集药:硫酸氢氯比格雷中间体的合成。 法国Sanofi-Aventi公司 (1998,美国)
选择性阻断ADP与血小板受体的结合。动脉粥样硬化,极性冠状动脉综合症。 2-(thiophen-3-yl)ethanamine
ethanamine.")
22
Example 细胞色素P450 抑制剂: 司替戊醇(Stiripentol,1) 的合成,化学名为4,4-二甲基-1-[(3,4-亚甲二氧基) 苯基]-1-戊烯-3-醇。 法国Biocodex 公司,欧盟首次上市(2007)。 临床用于治疗各种癫痫发作。 赵杰,张恺,华圆,程诚,杜玉民,司替戊醇的合成中国医药工业杂志 Chinese Journal of Pharmaceuticals 2013, 44(8) ·
![Example 细胞色素P450 抑制剂: 司替戊醇(Stiripentol,1) 的合成,化学名为4,4-二甲基-1-[(3,4-亚甲二氧基) 苯基]-1-戊烯-3-醇。 法国Biocodex 公司,欧盟首次上市(2007)。](http://slidesplayer.com/slide/11130619/59/images/22/Example+%E7%BB%86%E8%83%9E%E8%89%B2%E7%B4%A0P450+%E6%8A%91%E5%88%B6%E5%89%82%3A+%E5%8F%B8%E6%9B%BF%E6%88%8A%E9%86%87%28Stiripentol%EF%BC%8C1%29+%E7%9A%84%E5%90%88%E6%88%90%EF%BC%8C%E5%8C%96%E5%AD%A6%E5%90%8D%E4%B8%BA4%2C4-%E4%BA%8C%E7%94%B2%E5%9F%BA-1-%5B%283%2C4-%E4%BA%9A%E7%94%B2%E4%BA%8C%E6%B0%A7%E5%9F%BA%29+%E8%8B%AF%E5%9F%BA%5D-1-%E6%88%8A%E7%83%AF-3-%E9%86%87%E3%80%82+%E6%B3%95%E5%9B%BDBiocodex+%E5%85%AC%E5%8F%B8%EF%BC%8C%E6%AC%A7%E7%9B%9F%E9%A6%96%E6%AC%A1%E4%B8%8A%E5%B8%82%282007%29%E3%80%82.jpg "临床用于治疗各种癫痫发作。 赵杰,张恺,华圆,程诚,杜玉民,司替戊醇的合成中国医药工业杂志 Chinese Journal of Pharmaceuticals 2013, 44(8) ·")
23
Example 厄洛替尼(Erlotinib)中间体的合成
厄洛替尼,它塞瓦®(Tarceva®) 罗氏、基因技术及OSI制药公司(2007) Reduction:Text book(2015):p34(从化合物1-3-5到化合物1-3-6)
 罗氏、基因技术及OSI制药公司(2007) Reduction:Text book(2015):p34(从化合物1-3-5到化合物1-3-6)")
24
3 电子转移反应 又称为“溶解金属还原”(‘dissolving metal reduction’),以前认为涉及“初生态”氢。该反应涉及电子从金属原子上转移到反应底物上,使用金属包括:锂、钠、钾、镁、钙、锌、锡或铁。质子给予体(如:水或乙醇)既可存在于在电子转移期间,也可在以后阶段加入。羰基还原可形成三种类型产物,这要取决于反应条件[反应(8.2)]。还原成醇的反应在质子给予体存在下进行,开始形成的负离子自由基3首先被质子化,然后被第二个电子转移转化为碳负离子4。若没有质子给予体时,负离子自由基3则二聚为频哪醇盐双负离子5。克莱门森(Clemmensen)过程会涉及到向吸附在金属表面上的质子化的酮进行连续的电子转移。此时,为减少双分子还原,需在金属表面上保持较低浓度酮。
,以前认为涉及 初生态 氢。该反应涉及电子从金属原子上转移到反应底物上,使用金属包括:锂、钠、钾、镁、钙、锌、锡或铁。质子给予体(如:水或乙醇)既可存在于在电子转移期间,也可在以后阶段加入。羰基还原可形成三种类型产物,这要取决于反应条件[反应(8.2)]。还原成醇的反应在质子给予体存在下进行,开始形成的负离子自由基3首先被质子化,然后被第二个电子转移转化为碳负离子4。若没有质子给予体时,负离子自由基3则二聚为频哪醇盐双负离子5。克莱门森(Clemmensen)过程会涉及到向吸附在金属表面上的质子化的酮进行连续的电子转移。此时,为减少双分子还原,需在金属表面上保持较低浓度酮。")
25
Example 厄洛替尼(Erlotinib)中间体的合成
厄洛替尼,它塞瓦®(Tarceva®) 罗氏、基因技术及OSI制药公司(2007) Reduction:Text book(2015):p32
 罗氏、基因技术及OSI制药公司(2007) Reduction:Text book(2015):p32.")
26
Example 厄洛替尼(Erlotinib)中间体的合成
厄洛替尼,它塞瓦®(Tarceva®) 罗氏、基因技术及OSI制药公司(2007) Reduction:Text book(2015):p38(从化合物1-3-20到化合物1-3-21)
 罗氏、基因技术及OSI制药公司(2007) Reduction:Text book(2015):p38(从化合物1-3-20到化合物1-3-21)")
27
电化学方法和碘化钐(II)(SmI2) 电子转移反应还可通过电化学方法或使用低价态金属化合物实现。后者中最容易挥发的是碘化钐(II)(SmI2),由钐和二碘甲烷或1,2-二碘乙烷很方便制得。尽管该化合物对潮湿环境很敏感,它也可从商业直接购买。采用这种试剂,我们可完成许多涉及到含有卤素原子和氧原子的底物的官能团转化和偶合反应。
(SmI2),由钐和二碘甲烷或1,2-二碘乙烷很方便制得。尽管该化合物对潮湿环境很敏感,它也可从商业直接购买。采用这种试剂,我们可完成许多涉及到含有卤素原子和氧原子的底物的官能团转化和偶合反应。")
28
4 特殊官能团的还原

29
4.1 烯烃的还原反应 烯烃在催化剂下可迅速氢化生成烷烃,催化剂常是铂、雷尼镍、或在碳上的钯、铑。虽然常认为这种反应是一种立体专一顺式加成,但在催化剂表面所发生的重排过程却使这一结论显得过分简单化。 例如,1, 2-二甲基环己烯(6)异构化生成2, 3-二甲基环己烯(7),而化合物6催化氢化却得到顺式化合物8和反式化合物9(二甲基环己烷)的混合物。进行催化氘化时,这种异构化常会形成更复杂的混合物。
异构化生成2, 3-二甲基环己烯(7),而化合物6催化氢化却得到顺式化合物8和反式化合物9(二甲基环己烷)的混合物。进行催化氘化时,这种异构化常会形成更复杂的混合物。")
30
Example: 选择性褪黑激素受体激动剂:雷美替胺(Ramelteon) 的中间体合成。 日本武田制药株式会社(2005美国首次上市)。
治疗失眠症。 3-(2,3-dihydrobenzofuran-5-yl)propanoic acid
propanoic acid.")
31
改进:补充立体选择性还原试剂 三叔丁基硼氢化钠等的应用!

32
4.2 炔烃的还原反应 炔烃的催化氢化通过顺式(Z-)烯烃的过程还原而得到相应的烷烃。然而,如果使用活性被降低了的催化剂,如林德拉尔(Lindlar)催化剂(将附着在碳酸钡上的Pd经过乙酸铅处理并采用喹啉中毒化),林德拉尔催化剂对烯烃的吸附作用比对炔烃的吸附作用小,可以定量的产率获得顺式烯烃。其他令人满意的催化体系还可以通过对附着在硫酸钡上的Pd,以喹啉和镍(P-2)(通过采用NaBH4还原醋酸镍而得),在乙烯二胺的存在下进行部分中毒化而得。
烯烃的过程还原而得到相应的烷烃。然而,如果使用活性被降低了的催化剂,如林德拉尔(Lindlar)催化剂(将附着在碳酸钡上的Pd经过乙酸铅处理并采用喹啉中毒化),林德拉尔催化剂对烯烃的吸附作用比对炔烃的吸附作用小,可以定量的产率获得顺式烯烃。其他令人满意的催化体系还可以通过对附着在硫酸钡上的Pd,以喹啉和镍(P-2)(通过采用NaBH4还原醋酸镍而得),在乙烯二胺的存在下进行部分中毒化而得。")
33
4.3 醛和酮的还原反应

34
4.3.1 还原成醇

35
Meerwein-Ponndorf-Verley
在其他易被还原基团存在下还原羰基时,NPV很有用:

36
惰性溶剂/无水氧化铝/ 2-丙醇 优点: i) α, β-不饱和醛被还原成烯丙基醇; ii) 醛可在一些酮的存在下被还原;
iv) 丙醇/氧化铝可在一密闭小瓶中长期贮存; v) 试剂成本低且产物容易分离。
 丙醇/氧化铝可在一密闭小瓶中长期贮存; v) 试剂成本低且产物容易分离。")
37
提高选择性的其他方法 将α,β-不饱和醛和酮还原为烯丙基醇的选择性: 还可使用硼氢化钠和三氯化铈(III)甲醇溶液获得;
甲醇溶液获得;")
38
提高立体化学选择性的方法 涉及立体化学因素时d 情况更复杂。若使用氢化物还原可得两种差向异构体,其结果取决于以下两种因素:(i) 两种产物的相对稳定性,或 (ii) 氢化物试剂优先选择的进攻方向。当氢化物体积较大时,后者影响占主导地位,亲核试剂则从分子立体位阻较小的一侧进攻。当对氢化物试剂的立体因素并不做要求时,反应通常是生成更加稳定醇,并为优势产物。在电化学和溶解金属还原同样遵循后一种模式。 从分子位阻较小的一边进行催化氢化可得顺式(cis)加成产物。一般来讲正常情况下,镍和铹催化剂比较铂和钯的选择性较高。
 两种产物的相对稳定性,或 (ii) 氢化物试剂优先选择的进攻方向。当氢化物体积较大时,后者影响占主导地位,亲核试剂则从分子立体位阻较小的一侧进攻。当对氢化物试剂的立体因素并不做要求时,反应通常是生成更加稳定醇,并为优势产物。在电化学和溶解金属还原同样遵循后一种模式。 从分子位阻较小的一边进行催化氢化可得顺式(cis)加成产物。一般来讲正常情况下,镍和铹催化剂比较铂和钯的选择性较高。")
39
4-叔丁基环己酮的还原 4-叔丁基环己酮(14)在各种不同条件下被还原所形成的产物列于表4中,说明当需要考虑立体化学因素时,选择试剂的重要性。 化合物15和化合物16的相对热力学稳定性之比大约是4:1
在各种不同条件下被还原所形成的产物列于表4中,说明当需要考虑立体化学因素时,选择试剂的重要性。 化合物15和化合物16的相对热力学稳定性之比大约是4:1.")
40
表4 4-叔丁基环己酮的还原产物 还原剂 15:16的比率 LiAlH4 9:1 H2/雷尼镍/乙醇,1个大气压,20℃ 1:3
LiAl[OC(CH3)3]3/ THF H2/Rh/C/乙醇,1个大气压,20℃ 1:13 Li/在乙醚中的液氨/丁醇 49:1 Al[OCH(CH3)2]3 3:1
3]3/ THF. H2/Rh/C/乙醇,1个大气压,20℃ 1:13. Li/在乙醚中的液氨/丁醇. 49:1. Al[OCH(CH3)2]3. 3:1.")
41
3,3,5-三甲基环己酮的还原 3,3,5-三甲基环己酮(17)在各种不同条件下被还原所形成的产物列于表5中,目的还是用来说明当需要考虑立体化学因素时,选择试剂的重要性。 而化合物18和化合物19的相对热力学稳定性之比则大约是16:1
在各种不同条件下被还原所形成的产物列于表5中,目的还是用来说明当需要考虑立体化学因素时,选择试剂的重要性。 而化合物18和化合物19的相对热力学稳定性之比则大约是16:1.")
42
表5 3,3,5-三甲基环己酮还原产物 还原剂 18:19的比率 LiAlH4/ Et2O 1:1 Pt阴极/LiCl 电化学方法 10:1
LiAl[OC(CH3)3]3/ THF 1:8 Li/液氨/乙醇 99:1 H2/雷尼镍/乙醇,1个大气压,20℃ 1:32
3]3/ THF. 1:8. Li/液氨/乙醇. 99:1. H2/雷尼镍/乙醇,1个大气压,20℃ 1:32.")
43
4.3.2 双分子还原反应 当没质子给予体存在,酮和镁、锌或铝(常用作汞齐)反应时,最初形成的自由基离子发生二聚生成1,2-二醇(片呐醇)双负离子。双分子还原可与其他还原竞争,e.g.:Clemmensen reaction
反应时,最初形成的自由基离子发生二聚生成1,2-二醇(片呐醇)双负离子。双分子还原可与其他还原竞争,e.g.:Clemmensen reaction.")
44
其他试剂的使用 双分子还原还可通过使用各种试剂进行,包括“镁碳”(可通过碳化钾,C8K,和氯化镁反应制备) 和“低价钛”(可通过还原氯化钛(IV)制备而得)。多数情况下,产率较高,副产物较少。碘化亚钐(II)还可得双分子还原产物,在这种情况下,立体控制的获得是由于在中间体——钐羰游基自由基20中与Sm(III)的配位作用。这样的反应常可以导致环合。
 和 低价钛 (可通过还原氯化钛(IV)制备而得)。多数情况下,产率较高,副产物较少。碘化亚钐(II)还可得双分子还原产物,在这种情况下,立体控制的获得是由于在中间体——钐羰游基自由基20中与Sm(III)的配位作用。这样的反应常可以导致环合。")
45
4.3.3 酮还原成亚甲基 在无机酸存在下,锌汞齐反应可将羰基还原成亚甲基。该反应常在甲苯中进行,产生三相体系,大部分酮在上面有机烃层,存在于水层中的质子化羰基化合物则在金属表面上按下面机理被还原: Clemmensen reaction 在三相体系在金属表面上保持低浓度质子化羰基化合物减少双分子还原反应。

46
黄鸣龙(Huang-Minlon)法 对于克莱门森还原的补充反应就是用强碱来处理酮腙化合物的沃尔夫-基希涅尔反应(Wolff-Kishner reaction) (反应8.7)。改良方法包括黄鸣龙(Huang-Minlon)法:是将羰基化合物、水合肼和氢氧化钾混在一起在高沸点溶剂中加热。二甲基亚砜可使反应发生温度明显降低,至于对那些碱敏感的化合物,可选择另一方法,该法涉及酮基化合物的甲苯磺酰腙与氰基硼氢化钠反应。由于在这种条件下羰基化合物本身还原速度较慢,不需要预先将其生成腙。
 (反应8.7)。改良方法包括黄鸣龙(Huang-Minlon)法:是将羰基化合物、水合肼和氢氧化钾混在一起在高沸点溶剂中加热。二甲基亚砜可使反应发生温度明显降低,至于对那些碱敏感的化合物,可选择另一方法,该法涉及酮基化合物的甲苯磺酰腙与氰基硼氢化钠反应。由于在这种条件下羰基化合物本身还原速度较慢,不需要预先将其生成腙。")
47
其他补充方法 二硫代缩酮的氢解反应提供一种非常温和地把羰基转化为亚甲基的过程。
然而,因为该反应需要大大过量的雷尼镍(每克反应物需要7克雷尼镍), 所以该方法的应用通常仅仅局限于小规模的制备反应。
, 所以该方法的应用通常仅仅局限于小规模的制备反应。")
48
4.3.4 与羰基的还原偶合 我们将有关这种类型的碳-碳键的形成反应放在这一节讨论比放在第4.2节中讨论更为合适,因为尽管该反应的产物与格氏反应所形成的产物类似,但该反应过程并没有形成有机金属试剂。 羰基化合物与碘化钐(II)反应涉及到钐(III)-酮基自由基(21),其它各种官能团,如被-I和C=C等,可以将钐(III)-酮基自由基(21)俘获,然后最终生成产物,该方法用途最多的地方是在环合反应中。
反应涉及到钐(III)-酮基自由基(21),其它各种官能团,如被-I和C=C等,可以将钐(III)-酮基自由基(21)俘获,然后最终生成产物,该方法用途最多的地方是在环合反应中。")
49
碘化钐(II) 在碘化钐(II)的存在下,酮与卤代烷烃反应可以得到三级醇,这种产物的类型与格氏反应的产物结构相类似;醛发生反应方式与之类似,得到二级醇,但它只和活性卤代物反应(如:烯丙基卤代物和苄基卤代物)。其中具有更为重要意义的用途是环合反应,例如:
 在碘化钐(II)的存在下,酮与卤代烷烃反应可以得到三级醇,这种产物的类型与格氏反应的产物结构相类似;醛发生反应方式与之类似,得到二级醇,但它只和活性卤代物反应(如:烯丙基卤代物和苄基卤代物)。其中具有更为重要意义的用途是环合反应,例如:")
50
六甲基磷酸三酰胺(HMPA) 加入HMPA常可提高碘化钐(II)反应产率。人们相信HMPA和中间体20的络合延长了它的存在时间。
只有当烯烃被活化时,才可成功实现酮-烯烃分子间的偶合反应。

51
分子内的反应 分子内的反应不需要这样的活化,而在中等大小的环的形成中具有特殊的意义。

52
4.4 羧酸及其衍生物的还原反应 羧酸、酰胺和酯对催化氢化反应来讲是惰性的。事实上,乙酸乙酯和乙酸确实通常用作低压氢化溶剂。
4.4 羧酸及其衍生物的还原反应 羧酸、酰胺和酯对催化氢化反应来讲是惰性的。事实上,乙酸乙酯和乙酸确实通常用作低压氢化溶剂。 酯容易被氢化铝锂和溶解金属反应还原成醇。后者称为布沃-布朗方法(Bouveault-Blanc method),其优于氢化铝锂之处几乎寥寥无几,因而,布沃-布朗方法已经基本被氢化铝锂的方法所代替。羧酸也可被氢化铝锂还原生成伯醇。二元羧酸酯的偶姻反应已在第7.1.5节中讨论过。
,其优于氢化铝锂之处几乎寥寥无几,因而,布沃-布朗方法已经基本被氢化铝锂的方法所代替。羧酸也可被氢化铝锂还原生成伯醇。二元羧酸酯的偶姻反应已在第7.1.5节中讨论过。")
53
Adjusting Rosenmund catalyst

54
用金属氢化物还原羧酸衍生物制备醛 1. 从咪唑、咔唑或氮丙啶衍生得到的酰胺与氢化铝锂作用而被还原得到醛; 2.单叔酰胺与三乙氧基氢化铝锂
作用被还原可得醛.

55
制备醛的其他方法 3. 苯基酯则可采用三叔丁醇氢化铝锂进行还原得醛; 4. 而乙基羧酸酯在低温下与二异丁基氢化铝反应可得醛。

56
Mcfadyen-Stevens reaction
5. 羧酸也可通过磺酰肼反应转化生成醛,该反应与沃尔夫-基希涅尔反应相似,然而产率不高。 麦克法迪恩-史蒂文斯反应(Mcfadyen-Stevens reaction)
")
57
4.5 腈的还原反应 催化氢化还原和氢化铝锂还原都可以将腈基化合物转化成伯胺,虽然在前一种情况下产物会被仲胺杂质所沾污。认为反应中间体是亚胺20 ,如果还原反应可停留在这一步,则进一步水解反应将会形成醛。例如:

58
4.6 亚胺和肟的还原反应,包括还原烷基化 将亚胺催化氢化可以生成胺。与此密切相关的反应则是胺(包括氨)和硝基化合物的还原烷基化反应,最终将会导致形成伯胺、仲胺和叔胺。
和硝基化合物的还原烷基化反应,最终将会导致形成伯胺、仲胺和叔胺。")
59
氢化铝锂和硼氢化钠 亚胺和亚胺盐通过金属氢化物还原成胺时需要中性或稍为酸性的条件,使用的金属氢化物如氢化铝锂和硼氢化钠。在这种条件下,氰基硼氢化物的较高稳定性使它比其他种类的络合氢化物更适于实现这个转化反应。 在pH=6时,醛和酮仅能够被氰基硼氢化钠缓慢地还原,因此,还原烷基化作用在此条件下可以进行。

60
贝克曼重排 在乙酸介质中肟通过在铂上的催化氢化或使用溶解金属还原作用(如钠溶在乙醇中)可被还原成伯胺。采用氢化铝锂还原从脂肪肟几乎可专一地得伯胺。然而,相应芳基酮肟还原导致生成相当数量仲胺。如果该反应是在氯化铝存在的条件下进行,仲胺则可成为单一产物,大概是由于肟最初发生贝克曼重排:
可被还原成伯胺。采用氢化铝锂还原从脂肪肟几乎可专一地得伯胺。然而,相应芳基酮肟还原导致生成相当数量仲胺。如果该反应是在氯化铝存在的条件下进行,仲胺则可成为单一产物,大概是由于肟最初发生贝克曼重排:")
61
5 碳-杂原子键的还原断裂 人们通常将单键通过催化氢化的还原断裂称为氢解。卤化物进行氢解的难易程度取决于卤化物的类型(烷基卤化物较烯丙基,芳基,苄基和乙烯基卤化物难以被氢解)、卤素的种类(F<<Cl< Br < I)和催化剂的类型(钯催化剂比雷尼镍更有效,如果反应不需要氢解,则应该选择雷尼镍作为催化剂)以及溶剂的极性(极性溶剂和碱的存在有利于氢解)。因此,卤代苯胺和卤代吡啶在非酸性条件下很容易被氢解:
、卤素的种类(F<<Cl< Br < I)和催化剂的类型(钯催化剂比雷尼镍更有效,如果反应不需要氢解,则应该选择雷尼镍作为催化剂)以及溶剂的极性(极性溶剂和碱的存在有利于氢解)。因此,卤代苯胺和卤代吡啶在非酸性条件下很容易被氢解:")
62
氢化铝锂和硼氢化钠都可把一级卤代烷烃和二级卤代烷烃还原而生成烷烃。然而,在分子中共存的其他多种类型的官能团也会受到影响,这个反应看起来是涉及了在反应中心发生构型反转的SN2机理。在pH值大约等于6时,除碳-卤键之外,氰基硼氢化钠几乎不还原其他种类的官能团,因而,对于实现这个转化反应来讲,氰基硼氢化钠是一种高度专一性的试剂:

63
注意到卤代烷仅缓慢地被亲电的还原试剂——氢化铝进攻,这一点也是耐人寻味的,因此,在对其他官能团的还原过程中(23 → 24),选择这种试剂将会减少不需要的碳-卤键的断裂。
将氰基硼氢化物方法的推广和延伸则,可以为伯醇直接转变成烷烃化合物提供了另外一种方法:通过对中间体碘化物的分离并未使得产率得以改善。(仲醇和叔醇将会发生消除反应):
:")
64
卤代芳烃与氢化铝锂仅发生缓慢的反应,但有机锡氢化物则按照自由基的过程进攻卤化物,并可用来断裂卤代芳烃和其他不能发生SN2反应的卤代物:

65
对于易得的偕碳二卤代环丙烷的还原也具有重要的合成意义(参见第7. 2
对于易得的偕碳二卤代环丙烷的还原也具有重要的合成意义(参见第7.2.3节)。环丙烷的还原可以通过采用雷尼镍试剂或电子转移的方法完成,在后一种方法中使用的试剂有:在液氨或甲醇中的钠。三丁基锡氢化物是形成卤代环丙烷首选的方法,但是该法获得的却是异构体的混合物。通过电化学还原所得产物的比例介于1.6:1和5.3:1之间,具体比例要取决于所用溶剂的类型。
。环丙烷的还原可以通过采用雷尼镍试剂或电子转移的方法完成,在后一种方法中使用的试剂有:在液氨或甲醇中的钠。三丁基锡氢化物是形成卤代环丙烷首选的方法,但是该法获得的却是异构体的混合物。通过电化学还原所得产物的比例介于1.6:1和5.3:1之间,具体比例要取决于所用溶剂的类型。")
66
对于卤素以外的杂原子,合成上最重要的还原断裂则发生在苄基位置上。这可通过催化氢化的方法,或使用络合的金属氢化物或着采用电子转移方法来实现。氢解是通常可选择的方法,反应性能的顺序是:。这样就使苄基对于保护羟基和氨基显得非常有用(参见第10章)。
。")
67
其他重要的还原断裂反应 对于酰氯的罗森蒙德还原(参见第8.4.4节)和已经讨论过的使用雷尼镍(第8.4.3节)的脱硫作用,以及伯醇和仲醇经磺酸酯的还原作用。
和已经讨论过的使用雷尼镍(第8.4.3节)的脱硫作用,以及伯醇和仲醇经磺酸酯的还原作用。")
68
碘化钐(II) 碘化钐(II)可用于还原卤代烷烃,尤其是当被HMPA所活化的时候。许多α-取代酮很容易转化为未取代的酮,但是更重要的是一些环氧化物的开环和将羰基化合物的转化为多一个碳原子氰基同系物,其中α-氰基磷酸酯被还原。
 碘化钐(II)可用于还原卤代烷烃,尤其是当被HMPA所活化的时候。许多α-取代酮很容易转化为未取代的酮,但是更重要的是一些环氧化物的开环和将羰基化合物的转化为多一个碳原子氰基同系物,其中α-氰基磷酸酯被还原。")
69
6 环氧化物的还原开环 环氧乙烷衍生物(环氧化物)的两个C-O键都可以断裂,构型可以发生转化也可不发生转变,因此其氢解过程和产物较为复杂,下面的观察结果已被用在环氧化物的氢解中: (i) 在酸性溶剂中,氢化反应在铂催化剂上迅速发生,得到由更加稳定的碳正离子所衍生的开环产物:
的两个C-O键都可以断裂,构型可以发生转化也可不发生转变,因此其氢解过程和产物较为复杂,下面的观察结果已被用在环氧化物的氢解中: (i) 在酸性溶剂中,氢化反应在铂催化剂上迅速发生,得到由更加稳定的碳正离子所衍生的开环产物:")
70
(ii) 在碳上的钯是最有效的催化剂。并且在中性介质中,氢解发生在位阻较小C-O键上,得到取代基较多的醇:
 在碳上的钯是最有效的催化剂。并且在中性介质中,氢解发生在位阻较小C-O键上,得到取代基较多的醇:")
71
(iii) 用雷尼镍需要高压和高温,而在中性溶液中,取代基较少的醇占优势产物,但在碱的存在下则形成取代基较多的醇:
 用雷尼镍需要高压和高温,而在中性溶液中,取代基较少的醇占优势产物,但在碱的存在下则形成取代基较多的醇:")
72
(iv) 1-芳基-1,2-环氧化物在任何条件下开环都会得到1-芳基-2-羟基化合物。
使用氢化铝锂的还原,正如由SN2过程所预期的那样,通常导致环氧化物在较少取代的碳上 (如果可能的化则为伯碳)开环,得到取代基较多的醇。
开环,得到取代基较多的醇。")
73
使用亲电的氢化物试剂,开环的发生部位则倾向于有利于形成更稳定碳正离子,因而得到取代基较少的醇。重排还可以发生,这正如通过化合物27的形成时所看到的那样。

74
采用溶在乙二胺中的锂对环氧化物进行的还原开环反应也可导致生成取代基较多的醇,并且这对于有位阻的环氧化物来说是一种较具优势的方法。
采用碘化钐对环氧化物脱氧的一般性通用规则,就是应该注意到涉及适当取代的环氧化物的两种合成方法步骤。一种情况下,通过α, β-环氧基酮的还原开环可以形成醇醛类型的产物。如果手性环氧化物是由烯丙基醇的不对称环氧化而形成,继而转化为具有手性的α, β-环氧基酮,则可以由此合成具有手性的醇醛产物。

75
反式(E)-烯丙基醇可以通过烯丙基环氧乙烷开环而获得。如果烯丙基环氧化物本身具有手性,则可以再次得到手性产物。
-烯丙基醇可以通过烯丙基环氧乙烷开环而获得。如果烯丙基环氧化物本身具有手性,则可以再次得到手性产物。")
76
7α, β-不饱和羰基化合物的还原 碳-碳双键比羰基双键或腈基更容易发生氢化反应。在这种情况中,钯是我们优先选用的催化剂,而且,在碱性介质中,与羰基共轭的双键的氢化反应优先于孤立的双键的氢化反应。然而,对于这种氢化的立体化学的预测并非常常是可以容易进行的:

77
虽然,在低温下人们已经把这种反转加成的技术(即氢化物对α, β-不饱和化合物溶液的加成)成功用于将这类化合物还原成烯丙基醇,但是,对于实现这种转变的另一个更令人满意的试剂则是二异丁基氢化铝:
成功用于将这类化合物还原成烯丙基醇,但是,对于实现这种转变的另一个更令人满意的试剂则是二异丁基氢化铝:")
78
另一方面,溶解金属还原作用反应可导致碳-碳双键的还原,然而,产物的立体化学可能与催化氢化产物的立体化学不一致:

79
要将α,β-不饱和酮还原生成烯,通常是经过二硫缩酮的中间体,然后被雷尼镍脱硫而实现的,因为沃尔夫-基希涅尔还原过程会经过吡唑杂环31而将最终导致形成环丙烷,而克莱门森还原反应则得到较为复杂的混合物。

80
8 共轭二烯的还原 溶解金属还原1,3-二烯可导致1,4-加成,得到反式(E-)和顺式(Z-)烯烃的混和物,异构体的比率与温度有关。捕集实验表明,初期的自由基负离子(32)具有如下的顺式(Z-)构型: 共轭二烯在使用镍、铂或钯催化剂时可完全被氢化。 丁二烯的部分氢化的分析显示出1-丁烯, 反式(E)-2-丁烯和-顺式(Z)-2-丁烯分别以一定数量存在, 具体的物质的量的比例取决于所使用的催化剂种类。
-2-丁烯和-顺式(Z)-2-丁烯分别以一定数量存在, 具体的物质的量的比例取决于所使用的催化剂种类。")
81
9 芳香和杂芳香化合物的还原 苯环型化合物的化氢化反应常需高压条件,而在这种环境下,其他官能团比如烯烃的双键和羰基也同样会被还原。
苯环型的化合物对于氢化物试剂的还原一般不会受到影响,除非是分子中具有一个或一个以上的吸电子取代基时,将会受到一定的影响。 苯环型化合物的还原反应:伯奇还原

82
Birch 还原反应

83
Arthur John Birch Australia organic chemist
1915~1995 It is fitting that the first original experiments performed by Arthur Birch - distillation of eucalyptus leaves - were done on equipment bought from the legacy of a grandmother who called him professors as a child. His family was very poor and only after winning a scholarship at the University of Sydney did he find himself at Oxford to complete a PhD. After a successful research career at Oxford and Cambridge, where he formulated a breakthrough procedure in synthetic organic chemistry now known as the Birch Reduction, he was appointed to the Chair of Organic Chemistry at the University of Sydney. One of the world's great masters in organic chemistry, Birch's research formed the foundation for the manufacture of antibiotic drugs. From he was a foundation Professor at the Research School of Chemistry at the ANU and a leading science administrator and policy adviser. He was President of the Australian Academy of Science and one of the founders of the Australian Science and Technology Council. Birch was made a companion of the Order of Australia. He died, still actively engaged in science and writing, in 1996. From University of Sydney to Oxford PhD

84
反应式和定义 芳香族化合物在醇的存在下在液氨中用钠(锂或钾)还原,生成非共轭1,4-环二烯的反应称为Birch还原反应。
还原,生成非共轭1,4-环二烯的反应称为Birch还原反应。")
85
Birch还原反应三要素 The Birch Reduction converts aromatic compounds having a benzenoid ring into a product, 1,4-cyclohexadienes, in which two hydrogen atoms have been attached on opposite ends of the molecule. It is the organic reduction of aromatic rings in liquid ammonia with sodium, lithium or potassium and an alcohol, such as ethanol and tert-butanol. This reaction is quite unlike catalytic hydrogenation, which usually reduces the aromatic ring all the way to a cyclohexane.

86
Birch还原反应底物延伸 杂环化合物,如吡啶、吡咯和呋喃,也在这种情况下也能被还原,但上面的取代基必须为吸电子基EWG。

87
Birch 还原反应底物的延伸 当芳香化合物被取代时,还原反应区域选择性取决于取代基性质:如果取代基是供电子体,还原速度比非取代的化合物慢,取代基可以在新产物的非还原部分找到。在吸电子取代基的情况下,结果是相反的。 共轭烯烃,α,β-不饱和羰基化合物、环内炔烃、和苯乙烯衍生物也是可以被还原的。 换句话说,在多个”独立的“取代芳香环的底物中,含有吸电子基的芳香环则优先被还原,而含有供电子基的芳香环则难以被还原或不被还原。 请:注意区别与取代的稠合芳环(如萘环)被还原结果的不一样,但原因是一样的,都是由于自由基机理所经历的中间体的稳定性所致。
被还原结果的不一样,但原因是一样的,都是由于自由基机理所经历的中间体的稳定性所致。")
88
Birch还原的局限性 普通烯烃不受Birch还原条件影响,双键若没与芳香环共轭则不被还原而存在。
富电子的杂环需要有至少含有一个吸电子的取代基(EWG)。 未取代的呋喃和噻吩不被还原,除非含有吸电子取代基。
。 未取代的呋喃和噻吩不被还原,除非含有吸电子取代基。")
89
Birch 还原反应的机理 Typical free radical mechanism

90
以钠为例分析机理: 请注意中间体自由基所在位置应该有利于中间体的稳定性。

91
Birch还原反应结果 在具有吸电子取代基的化合物中,还原反应发生在带有取代基的碳上。
If R=EWG then the product is: 1-取代-2,5-环己二烯 在具有给电子取代基的化合物中,还原反应则发生在其中一个邻位碳上。 If R=EDG then the product is: 1-取代-1,4-环己二烯

92
反应结果原因分析 中间体负离子自由基(33)和(34)的相对稳定性来推理解释。
和(34)的相对稳定性来推理解释。")
93
由此类推 在双环化合物中,电子云密度较低的环被还原。

94
在Na+EtOH条件下Bouveault-Blanc Reduction还原反应 和在醇钠(EtONa)催化条件下的Claisen缩合反应。
有关Birch 还原条件的知识联想 说明: 1. 醇可是乙醇、异丙醇或仲丁醇等。 2. 区分Na+NH3和NaNH2两个体系 Na+NH3提供电子,还原 NaNH2为强碱,夺氢 联想类比: 在Na+EtOH条件下Bouveault-Blanc Reduction还原反应 和在醇钠(EtONa)催化条件下的Claisen缩合反应。
催化条件下的Claisen缩合反应。")
95
Birch 还原的影响因素 当苯环上有取代基时: 阴离子自由基 EWG:COOH,EDG:Me

96
在药物合成中的应用 18-甲基炔诺酮(Norgestrel)中间体的合成:
The Birch Reduction enables the modification of steroids. In 1948 Birch published the first total synthesis of a male sex hormone (19-nortestosterone), as the first member of a new structural series. This series later comprised the first oral contraceptive pill, which was made by others. The Birch reduction also allows for the development of other steroid drugs and antibiotics The first simple synthesis of the ring A-B structure of cholesterol..
, as the first member of a new structural series. This series later comprised the first oral contraceptive pill, which was made by others. The Birch reduction also allows for the development of other steroid drugs and antibiotics. The first simple synthesis of the ring A-B structure of cholesterol..")
97
在有机合成中的应用 2. 取代环己酮衍生物的制备:
环己酮衍生物的合成可以用Birch还原反应制备,可能的原料为:取代苯环衍生物,如苯甲醚和苯胺类。

98
在有机合成中的应用 在第一个例子(I)中,T.J.Donohoe等人利用Birch还原反应将缺电子的2-和3-取代吡咯类化合物还原之后接着再进行烷基化反应。39,40这种还原烷基化的方法被证明是分别合成取代的3-和2-吡咯烷类化合物的非常有效的方法。作为一个质子源的醇对于还原反应的发生并非必需。在第二个例子(II)中,同样的研究者们在对(+)-Nemorensic酸的全合成过程中,对取代呋喃进行了一个立体选择性Birch还原反应。
中,T.J.Donohoe等人利用Birch还原反应将缺电子的2-和3-取代吡咯类化合物还原之后接着再进行烷基化反应。39,40这种还原烷基化的方法被证明是分别合成取代的3-和2-吡咯烷类化合物的非常有效的方法。作为一个质子源的醇对于还原反应的发生并非必需。在第二个例子(II)中,同样的研究者们在对(+)-Nemorensic酸的全合成过程中,对取代呋喃进行了一个立体选择性Birch还原反应。")
99
在有机合成中的应用 紫杉醇全合成 在对(–)-紫杉醇对映体选择性的合成过程中,I.Kuwajima及其同事们利用Birch还原反应精心构建了在天然产物的C-环上的官能团排列。42原始的1,2-二取代苯环经历了典型的Birch还原反应条件 (钾/液氨/叔丁醇),由此产生的1,3-环己二烯(I)被单线态的氧从凸起的β-面氧化形成所需的C4β-C7β二醇。副产物苄醇衍生物(II)可以通过Swern氧化反应被回收为起始原料,产率非常高,由此使得总转化率对于合成目的来说还是可以接受的。
-紫杉醇对映体选择性的合成过程中,I.Kuwajima及其同事们利用Birch还原反应精心构建了在天然产物的C-环上的官能团排列。42原始的1,2-二取代苯环经历了典型的Birch还原反应条件 (钾/液氨/叔丁醇),由此产生的1,3-环己二烯(I)被单线态的氧从凸起的β-面氧化形成所需的C4β-C7β二醇。副产物苄醇衍生物(II)可以通过Swern氧化反应被回收为起始原料,产率非常高,由此使得总转化率对于合成目的来说还是可以接受的。")
100
(+)-阿扑长春胺的合成 在A.G.Schultz的实验室中,在两个Vincane型的生物碱(+)-阿扑长春胺和(+)-长春胺的不对称全合成中,有必要构建一个关键的顺式稠合的五元环二烯中间体。43合成从对手性苯甲酰胺的Birch还原-烷基化反应开始,得到6-乙基-1-甲氧基-4-甲基-1,4-环己二烯的非对映体纯度比例大于100:1。这个环己二烯首先被转换为一个对映异构体纯的丁内酯,该化合物经过几步反应之后被转化为(+)-阿扑长春胺。
-阿扑长春胺和(+)-长春胺的不对称全合成中,有必要构建一个关键的顺式稠合的五元环二烯中间体。43合成从对手性苯甲酰胺的Birch还原-烷基化反应开始,得到6-乙基-1-甲氧基-4-甲基-1,4-环己二烯的非对映体纯度比例大于100:1。这个环己二烯首先被转换为一个对映异构体纯的丁内酯,该化合物经过几步反应之后被转化为(+)-阿扑长春胺。")
101
Galbulimima(白木兰属)生物碱GB 13的全合成
Galbulimima(白木兰属)生物碱GB 13的全合成是由L.N.Mander及其同事们完成的。为了制备环状α,β-不饱和酮需要对复杂中间体进行Birch还原反应。44将底物与锂金属在液氨中进行处理首次得到对C6a氰基的定量还原脱氰基化的结果。向反应混合物中加入过量的乙醇可将芳香环还原为相应的烯醇醚,该化合物接着水解得到不饱和酮。
生物碱GB 13的全合成是由L.N.Mander及其同事们完成的。为了制备环状α,β-不饱和酮需要对复杂中间体进行Birch还原反应。44将底物与锂金属在液氨中进行处理首次得到对C6a氰基的定量还原脱氰基化的结果。向反应混合物中加入过量的乙醇可将芳香环还原为相应的烯醇醚,该化合物接着水解得到不饱和酮。")
102
应用:合成1, 5-二羰基化合物 取代吡啶在伯奇还原的条件能得1, 4-二氢吡啶(环状烯胺),易水解而得1, 5-二羰基化合物:
相关知识点的类比: 吡啶鎓盐的高压氢化还原或使用硼氢化钠进行还原可导致哌啶形成;而根据反应条件,还可分离出来1, 2-二氢和1, 2, 5, 6-四氢吡啶。在质子给予体存在下,吡啶的溶解金属还原也可导致哌啶生成。 Cyclic enamine 1,5-dicarbonyl compound

103
延伸阅读 Enzymatic Birch Reduction
The Birch reduction is a widely used synthetic tool in organic chemistry that achieves 1,4-dihydro additions to benzenoid and other aromatic compounds. The reaction proceeds by alternate electron transfer and protonation steps to the aromatic ring and requires solvated electrons, which are usually generated by dissolving an alkali metal in liquid ammonia. Considering these nonphysiological conditions it is remarkable that a similar reaction exists in biology: the dearomatizing benzoyl-coenzyme A reductase (BCR) plays a key role in the anaerobic degradation of aromatic compounds.
 plays a key role in the anaerobic degradation of aromatic compounds.")
104
Dearomatizing benzoyl-coenzyme A reductase (BCR)
In anaerobic bacteria many low-molecular aromatic growth substrates are converted to the central intermediate benzoyl-CoA (BCoA), which serves as substrate for BCR. The enzyme catalyzes the reduction of BCoA (1) to cyclohexa-1,5-diene-1-carboxyl-CoA (dienoyl-CoA, 2) rather than the kinetically favored 2,5-dienoyl-CoA isomer.
, which serves as substrate for BCR. The enzyme catalyzes the reduction of BCoA (1) to cyclohexa-1,5-diene-1-carboxyl-CoA (dienoyl-CoA, 2) rather than the kinetically favored 2,5-dienoyl-CoA isomer.")
105
Enzymatic Mechanism A mechanism similar to the classical Birch reduction has been suggested in which the rate limiting first electron transfer yields a radical anion. The CoA ester moiety is considered to stabilize this intermediate by formation of a relatively stable thioester ketyl radical. Remarkably, BCR couples electron transfer to the aromatic ring from the donor reduced ferredoxin (Fd) to a stoichiometric ATP hydrolysis, a reaction that has long been considered as an exclusive feature of nitrogenase. The oxygen sensitive BCR has so far only been isolated from the facultatively anaerobic bacterium Thauera aromatica. Barbel Thiele, Oliver Rieder, Bernard T. Golding, Michael Mu ller, Matthias Boll Mechanism of Enzymatic Birch Reduction: Stereochemical Course and Exchange Reactions of Benzoyl-CoA Reductase,J. AM. CHEM. SOC. 2008, 130, 14050–14051.
 to a stoichiometric ATP hydrolysis, a reaction that has long been considered as an exclusive feature of nitrogenase. The oxygen sensitive BCR has so far only been isolated from the facultatively anaerobic bacterium Thauera aromatica. Barbel Thiele, Oliver Rieder, Bernard T. Golding, Michael Mu ller, Matthias Boll Mechanism of Enzymatic Birch Reduction: Stereochemical Course and Exchange Reactions of Benzoyl-CoA Reductase,J. AM. CHEM. SOC. 2008, 130, 14050–")
106
The end!

107
Summary 目前共认识到了四种类型的还原反应,包括:催化氢化反应(采用非均相和均相催化剂)、催化转移氢化反应、采用金属氢化物的还原反应以及电子转移的过程。本章还描述了以下几种还原方法,并给出例子,还讨论了立体选择性。 炔烃 烯烃 烷烃 C=O CH(OH) C=O CH2 (克莱门森还原,沃尔夫-基希涅尔还原) COCl CHO (罗森蒙德还原,麦克法迪恩还原) C≡N CHO C=N CH(NH) CH=NOH CH2NH2 水解 (R-X R-H) C=C-C=O CH-CH-C=O 或C=C-CH(OH) CH=CH-CH=CH CH2-CH=CH-CH2 (包括伯奇还原) 2 C=O C(OH)-C(OH)
 C=O CH2 (克莱门森还原,沃尔夫-基希涅尔还原) COCl CHO (罗森蒙德还原,麦克法迪恩还原) C≡N CHO. C=N CH(NH) CH=NOH CH2NH2. 水解 (R-X R-H) C=C-C=O CH-CH-C=O 或C=C-CH(OH) CH=CH-CH=CH CH2-CH=CH-CH2 (包括伯奇还原) 2 C=O C(OH)-C(OH)")
108
氧化反应 PART II Oxidation

109
氧化反应 2.1 一般性原则 2.2 烃的氧化反应 2.3 醇及其衍生物的氧化反应 2.4 芳香体系的氧化反应 2.5 醛和酮的氧化反应
2.1 一般性原则 2.2 烃的氧化反应 2.3 醇及其衍生物的氧化反应 2.4 芳香体系的氧化反应 2.5 醛和酮的氧化反应 2.6 含氮官能团的氧化反应 2.7 含硫官能团的氧化反应

110
Introduction 氧化反应就是还原反应对立面,因此,在本章反应从原理上是第8章所描述反应的逆过程。我们已介绍了三种不同类型的还原,即氢对重键的加成(使用催化的和非催化的方法);官能团被氢的取代和单电子对亲电中心的加成。因此,这些过程的对立面则应当可以成为氧化反应,即消除氢以形成重键;氢被官能团取代和从亲核中心夺取一个电子。 正如下文中将会明显看到的那样,所有三种类型反应的例子都是人们所熟知的。在这三种反应类型中必须再加上一种,即第四种,就是含氧试剂对重键的加成(一般说来,与这种反应相互对映的还原反应很少见,因此,在第8章中没有考虑)和含氧试剂与杂原子的加成,如氮、磷和硫等杂原子。
;官能团被氢的取代和单电子对亲电中心的加成。因此,这些过程的对立面则应当可以成为氧化反应,即消除氢以形成重键;氢被官能团取代和从亲核中心夺取一个电子。 正如下文中将会明显看到的那样,所有三种类型反应的例子都是人们所熟知的。在这三种反应类型中必须再加上一种,即第四种,就是含氧试剂对重键的加成(一般说来,与这种反应相互对映的还原反应很少见,因此,在第8章中没有考虑)和含氧试剂与杂原子的加成,如氮、磷和硫等杂原子。")
111
2.1 一般性原则 2.1.1 脱氢反应 (i) 催化脱氢. (ii) 通过连续的氢负离子和质子转移脱氢.
2.1 一般性原则 2.1.1 脱氢反应 (i) 催化脱氢. (ii) 通过连续的氢负离子和质子转移脱氢. (iii) 通过取代-消除和加成-消除过程脱氢.
 催化脱氢. (ii) 通过连续的氢负离子和质子转移脱氢. (iii) 通过取代-消除和加成-消除过程脱氢.")
112
2.1.2 氢被一个官能团取代 氢解(官能团被氢原子所取代)的对立面就是官能化作用,这一主题已经在第2章中作以简略讨论。然而,把一般的官能团化反应看作为氧化过程却是非同寻常的,除非是氢原子被氧化性官能团所取代,比如被OH取代之外。这些反应发生的机理既可以是自由基的也以是离子型的[反应(9.6)和反应(9.7)],例如:
的对立面就是官能化作用,这一主题已经在第2章中作以简略讨论。然而,把一般的官能团化反应看作为氧化过程却是非同寻常的,除非是氢原子被氧化性官能团所取代,比如被OH取代之外。这些反应发生的机理既可以是自由基的也以是离子型的[反应(9.6)和反应(9.7)],例如:")
113
2.1.3 从亲核中心抽取单电子 这种反应最普通形式包括从负离子中抽取一个电子而得到自由基,接着发生自由基的二聚化作用[反应(9.8)]。这样的过程早在第4章中有关有机铜衍生物的内容里,我们已经遇到(见第44页和第47页)。如果自由基因离域化而得到稳定,该自由基偶合则可得一个不对称二聚体(参见第9.4节):
]。这样的过程早在第4章中有关有机铜衍生物的内容里,我们已经遇到(见第44页和第47页)。如果自由基因离域化而得到稳定,该自由基偶合则可得一个不对称二聚体(参见第9.4节):")
114
2.1.4 含氧试剂对重键和杂原子加成反应 包括两种类型反应:多重键的羟基化反应[例如:反应(9.9)]和氧原子(通常是来自过氧酸)与含有孤对儿电子的杂原子之间的加成反应[反应(9.10)]:
![2.1.4 含氧试剂对重键和杂原子加成反应 包括两种类型反应:多重键的羟基化反应[例如:反应(9.9)]和氧原子(通常是来自过氧酸)与含有孤对儿电子的杂原子之间的加成反应[反应(9.10)]:](http://slidesplayer.com/slide/11130619/59/images/114/2.1.4+%E5%90%AB%E6%B0%A7%E8%AF%95%E5%89%82%E5%AF%B9%E9%87%8D%E9%94%AE%E5%92%8C%E6%9D%82%E5%8E%9F%E5%AD%90%E5%8A%A0%E6%88%90%E5%8F%8D%E5%BA%94+%E5%8C%85%E6%8B%AC%E4%B8%A4%E7%A7%8D%E7%B1%BB%E5%9E%8B%E5%8F%8D%E5%BA%94%EF%BC%9A%E5%A4%9A%E9%87%8D%E9%94%AE%E7%9A%84%E7%BE%9F%E5%9F%BA%E5%8C%96%E5%8F%8D%E5%BA%94%5B%E4%BE%8B%E5%A6%82%EF%BC%9A%E5%8F%8D%E5%BA%94%289.9%29%5D%E5%92%8C%E6%B0%A7%E5%8E%9F%E5%AD%90%28%E9%80%9A%E5%B8%B8%E6%98%AF%E6%9D%A5%E8%87%AA%E8%BF%87%E6%B0%A7%E9%85%B8%29%E4%B8%8E%E5%90%AB%E6%9C%89%E5%AD%A4%E5%AF%B9%E5%84%BF%E7%94%B5%E5%AD%90%E7%9A%84%E6%9D%82%E5%8E%9F%E5%AD%90%E4%B9%8B%E9%97%B4%E7%9A%84%E5%8A%A0%E6%88%90%E5%8F%8D%E5%BA%94%5B%E5%8F%8D%E5%BA%94%289.10%29%5D%EF%BC%9A.jpg "2.1.4 含氧试剂对重键和杂原子加成反应 包括两种类型反应:多重键的羟基化反应[例如:反应(9.9)]和氧原子(通常是来自过氧酸)与含有孤对儿电子的杂原子之间的加成反应[反应(9.10)]:")
115
2.2 烃的氧化反应 这当然是工业上感兴趣的极为重要的领域,它不仅与燃料有关,而且也与橡胶、塑料和粮食等方面有关。然而,本部分的目的却不是要讨论这些内容,而是从实验室的角度出发,集中讨论一些在合成上有用的氧化反应。

116
9.2.1 烷烃和烷基 像烷烃的其他反应一样,氧化反应按照自由基机理进行[反应(9.6)],在反应过程中抽取一个氢原子是反应第一步。如果氢原子的抽取仅能发生在一个特定位置,这就是一个有用的合成方法。例如,三级氢比二级氢或一级氢更容易被抽取走,一些支链烷烃因而可以被氧化成叔醇,如,
![9.2.1 烷烃和烷基 像烷烃的其他反应一样,氧化反应按照自由基机理进行[反应(9.6)],在反应过程中抽取一个氢原子是反应第一步。如果氢原子的抽取仅能发生在一个特定位置,这就是一个有用的合成方法。例如,三级氢比二级氢或一级氢更容易被抽取走,一些支链烷烃因而可以被氧化成叔醇,如,](http://slidesplayer.com/slide/11130619/59/images/116/9.2.1+%E7%83%B7%E7%83%83%E5%92%8C%E7%83%B7%E5%9F%BA+%E5%83%8F%E7%83%B7%E7%83%83%E7%9A%84%E5%85%B6%E4%BB%96%E5%8F%8D%E5%BA%94%E4%B8%80%E6%A0%B7%EF%BC%8C%E6%B0%A7%E5%8C%96%E5%8F%8D%E5%BA%94%E6%8C%89%E7%85%A7%E8%87%AA%E7%94%B1%E5%9F%BA%E6%9C%BA%E7%90%86%E8%BF%9B%E8%A1%8C%5B%E5%8F%8D%E5%BA%94%289.6%29%5D%EF%BC%8C%E5%9C%A8%E5%8F%8D%E5%BA%94%E8%BF%87%E7%A8%8B%E4%B8%AD%E6%8A%BD%E5%8F%96%E4%B8%80%E4%B8%AA%E6%B0%A2%E5%8E%9F%E5%AD%90%E6%98%AF%E5%8F%8D%E5%BA%94%E7%AC%AC%E4%B8%80%E6%AD%A5%E3%80%82%E5%A6%82%E6%9E%9C%E6%B0%A2%E5%8E%9F%E5%AD%90%E7%9A%84%E6%8A%BD%E5%8F%96%E4%BB%85%E8%83%BD%E5%8F%91%E7%94%9F%E5%9C%A8%E4%B8%80%E4%B8%AA%E7%89%B9%E5%AE%9A%E4%BD%8D%E7%BD%AE%EF%BC%8C%E8%BF%99%E5%B0%B1%E6%98%AF%E4%B8%80%E4%B8%AA%E6%9C%89%E7%94%A8%E7%9A%84%E5%90%88%E6%88%90%E6%96%B9%E6%B3%95%E3%80%82%E4%BE%8B%E5%A6%82%EF%BC%8C%E4%B8%89%E7%BA%A7%E6%B0%A2%E6%AF%94%E4%BA%8C%E7%BA%A7%E6%B0%A2%E6%88%96%E4%B8%80%E7%BA%A7%E6%B0%A2%E6%9B%B4%E5%AE%B9%E6%98%93%E8%A2%AB%E6%8A%BD%E5%8F%96%E8%B5%B0%EF%BC%8C%E4%B8%80%E4%BA%9B%E6%94%AF%E9%93%BE%E7%83%B7%E7%83%83%E5%9B%A0%E8%80%8C%E5%8F%AF%E4%BB%A5%E8%A2%AB%E6%B0%A7%E5%8C%96%E6%88%90%E5%8F%94%E9%86%87%EF%BC%8C%E5%A6%82%EF%BC%8C.jpg "9.2.1 烷烃和烷基 像烷烃的其他反应一样,氧化反应按照自由基机理进行[反应(9.6)],在反应过程中抽取一个氢原子是反应第一步。如果氢原子的抽取仅能发生在一个特定位置,这就是一个有用的合成方法。例如,三级氢比二级氢或一级氢更容易被抽取走,一些支链烷烃因而可以被氧化成叔醇,如,")
117
重排变化 然而,这个反应普遍比用格氏试剂的方法(第4.1.2节)制备第三醇要差一些。从合成的观点来看,更重要的是基团的氧化,其中,通过分子内的氢抽取而产生自由基;1式的自由基可能经过一个六员的过渡态重排成2式的自由基[反应(9.11)]:
![重排变化 然而,这个反应普遍比用格氏试剂的方法(第4.1.2节)制备第三醇要差一些。从合成的观点来看,更重要的是基团的氧化,其中,通过分子内的氢抽取而产生自由基;1式的自由基可能经过一个六员的过渡态重排成2式的自由基[反应(9.11)]:](http://slidesplayer.com/slide/11130619/59/images/117/%E9%87%8D%E6%8E%92%E5%8F%98%E5%8C%96+%E7%84%B6%E8%80%8C%EF%BC%8C%E8%BF%99%E4%B8%AA%E5%8F%8D%E5%BA%94%E6%99%AE%E9%81%8D%E6%AF%94%E7%94%A8%E6%A0%BC%E6%B0%8F%E8%AF%95%E5%89%82%E7%9A%84%E6%96%B9%E6%B3%95%28%E7%AC%AC4.1.2%E8%8A%82%29%E5%88%B6%E5%A4%87%E7%AC%AC%E4%B8%89%E9%86%87%E8%A6%81%E5%B7%AE%E4%B8%80%E4%BA%9B%E3%80%82%E4%BB%8E%E5%90%88%E6%88%90%E7%9A%84%E8%A7%82%E7%82%B9%E6%9D%A5%E7%9C%8B%EF%BC%8C%E6%9B%B4%E9%87%8D%E8%A6%81%E7%9A%84%E6%98%AF%E5%9F%BA%E5%9B%A2%E7%9A%84%E6%B0%A7%E5%8C%96%EF%BC%8C%E5%85%B6%E4%B8%AD%EF%BC%8C%E9%80%9A%E8%BF%87%E5%88%86%E5%AD%90%E5%86%85%E7%9A%84%E6%B0%A2%E6%8A%BD%E5%8F%96%E8%80%8C%E4%BA%A7%E7%94%9F%E8%87%AA%E7%94%B1%E5%9F%BA%EF%BC%9B1%E5%BC%8F%E7%9A%84%E8%87%AA%E7%94%B1%E5%9F%BA%E5%8F%AF%E8%83%BD%E7%BB%8F%E8%BF%87%E4%B8%80%E4%B8%AA%E5%85%AD%E5%91%98%E7%9A%84%E8%BF%87%E6%B8%A1%E6%80%81%E9%87%8D%E6%8E%92%E6%88%902%E5%BC%8F%E7%9A%84%E8%87%AA%E7%94%B1%E5%9F%BA%5B%E5%8F%8D%E5%BA%94%289.11%29%5D%EF%BC%9A.jpg "重排变化 然而,这个反应普遍比用格氏试剂的方法(第4.1.2节)制备第三醇要差一些。从合成的观点来看,更重要的是基团的氧化,其中,通过分子内的氢抽取而产生自由基;1式的自由基可能经过一个六员的过渡态重排成2式的自由基[反应(9.11)]:")
118
Barton reaction 巴顿反应(Barton reaction) 自由基是通过亚硝酸酯光解而产生的:
 自由基是通过亚硝酸酯光解而产生的:")
119
甾体化合物的合成 甾体化合物的分子内氢原子转移常被分子骨架刚性和相互作用的官能团之间的1,3-二直立键的关系这两点因素所促进(参见3)。
该反应成功用于甾体化合物的选择性氧化。 醛固酮(aldosterone, 4)
")
120
非环状的亚硝酸酯的巴顿反应 除了发生分子内氢反应之外,抽取像1这样的含氧自由基显然还能够发生其他类型反应,因此,可以预期,非环状的亚硝酸酯由于缺乏甾体骨架的刚性,在巴顿反应中可能会给出非常低的产率。然而实际上在某些情况下产率却还比较高,如:

121
9.2.2 烯丙位的氧化反应 从烯丙位上抽取氢原子比从完全饱和的烷烃上抽取氢原子更加容易,这是因为,生成的自由基可以因为共振效应而稳定。
9.2.2 烯丙位的氧化反应 从烯丙位上抽取氢原子比从完全饱和的烷烃上抽取氢原子更加容易,这是因为,生成的自由基可以因为共振效应而稳定。 同样,烯丙基正离子和负离子相对于其完全饱和的对映物来说也是稳定的, 因此,几种不同类型的烯丙位的氧化涉及烯丙自由基、正离子或负离子作为中间体理应是可能的。 当然,烯烃本身的氧化反应就是一种竞争过程,因而中间体本身在烯丙基体系的“两端”均可发生反应。 通过使用N-溴代丁二酰亚胺进行溴化,接着发生溴化物的水解来实现氧化生成醇也许是最简单的方式。

122
9.2.2 烯丙位的氧化反应 氧化的官能团还可以通过使用四醋酸铅(IV)、二氧化硒(IV)或过酸酯在一价铜(I)盐的存在下直接引入,如:
、二氧化硒(IV)或过酸酯在一价铜(I)盐的存在下直接引入,如:")
123
机理初步分析 需要注意第一个实例和第二个实例或许是包含了对最初双键的进攻和生成像5、6或7中间体。
第三个实例涉及烯丙基自由基,然后是烯丙基正离子[反应(9.12)]:
]:")
124
更强且选择性较小的氧化剂可使烯丙位氧化越过醇阶段,还能够引起双键氧化,如:

125
高锰酸钾 高锰酸钾对于烯丙位的氧化反应常常不会是令人满意的结果, 因为它将会优先与双键发生反应(参见第9.2.5节) 。
 。")
126
9.2.3 苄基位的氧化反应 上节注意到自由基、正离子和负离子被邻近双键的稳定作用,以及由这些烯丙位的氧化必然导出的多种多样的可能机理途径。苄基位氧化也存在着同样多样性,因为芳香体系可用来稳定自由基或苄基碳上的电荷,而涉及苄基自由基、正离子和负离子的氧化全都是已知的(参见以下反应)。 在这些反应中,在分子其他部位发生氧化,通常不会是一个严重问题,因为,芳香环一般说来较难被氧化。

127
强氧化剂 强氧化剂可使苄基碳原子氧化到最高价位,这些氧化剂包括高锰酸钾或铬酸,如:

128
剪发头反应 在这些条件下,取代烷基中含有两个或二个以上碳原子的其他烷基苯化合物也可以被氧化成苯甲酸类衍生物。人们认为最初的氧化反应是发生在苄基位上,因为叔丁基苯(它没有苄基氢原子)是耐氧化的,也由于芳基烷基酮偶尔也作为副产物而被分离出来。
是耐氧化的,也由于芳基烷基酮偶尔也作为副产物而被分离出来。")
129
温和氧化产物 在没有外加酸的存在下,使用含水的重铬酸钠作氧化剂则提供了稍为温和的条件,此时观察不到烷基的断裂,例如:

130
稠环芳香化合物的氧化产物 据试剂不同,稠环芳香化合物可生成不同产物。如,在酸性介质中,使用铬(VI)试剂可把萘氧化成萘醌,而没有酸时,重铬酸钠仅能够氧化取代基。
试剂可把萘氧化成萘醌,而没有酸时,重铬酸钠仅能够氧化取代基。")
131
高锰酸钾的开环 高锰酸钾可使氧化更进一步,能使环发生破裂同时形成单环二羧酸:

132
将苄基中心氧化到比可达到的最高氧化态的较低级的水平则显得更为困难。例如,反应ArCH3 ArCHO和反应ArCH2R ArCH(OH)R都是困难的,因为生成的产物比反应物更容易被发生氧化。已经提出了把甲基控制氧化成醛的几种方法。最简单的一种涉及甲基的自由基卤代和后续的分离以及二卤代衍生物的水解[反应(9.13)]:
![将苄基中心氧化到比可达到的最高氧化态的较低级的水平则显得更为困难。例如,反应ArCH3 ArCHO和反应ArCH2R ArCH(OH)R都是困难的,因为生成的产物比反应物更容易被发生氧化。已经提出了把甲基控制氧化成醛的几种方法。最简单的一种涉及甲基的自由基卤代和后续的分离以及二卤代衍生物的水解[反应(9.13)]:](http://slidesplayer.com/slide/11130619/59/images/132/%E5%B0%86%E8%8B%84%E5%9F%BA%E4%B8%AD%E5%BF%83%E6%B0%A7%E5%8C%96%E5%88%B0%E6%AF%94%E5%8F%AF%E8%BE%BE%E5%88%B0%E7%9A%84%E6%9C%80%E9%AB%98%E6%B0%A7%E5%8C%96%E6%80%81%E7%9A%84%E8%BE%83%E4%BD%8E%E7%BA%A7%E7%9A%84%E6%B0%B4%E5%B9%B3%E5%88%99%E6%98%BE%E5%BE%97%E6%9B%B4%E4%B8%BA%E5%9B%B0%E9%9A%BE%E3%80%82%E4%BE%8B%E5%A6%82%EF%BC%8C%E5%8F%8D%E5%BA%94ArCH3+%EF%80%A2+ArCHO%E5%92%8C%E5%8F%8D%E5%BA%94ArCH2R+%EF%80%A2+ArCH%28OH%29R%E9%83%BD%E6%98%AF%E5%9B%B0%E9%9A%BE%E7%9A%84%EF%BC%8C%E5%9B%A0%E4%B8%BA%E7%94%9F%E6%88%90%E7%9A%84%E4%BA%A7%E7%89%A9%E6%AF%94%E5%8F%8D%E5%BA%94%E7%89%A9%E6%9B%B4%E5%AE%B9%E6%98%93%E8%A2%AB%E5%8F%91%E7%94%9F%E6%B0%A7%E5%8C%96%E3%80%82%E5%B7%B2%E7%BB%8F%E6%8F%90%E5%87%BA%E4%BA%86%E6%8A%8A%E7%94%B2%E5%9F%BA%E6%8E%A7%E5%88%B6%E6%B0%A7%E5%8C%96%E6%88%90%E9%86%9B%E7%9A%84%E5%87%A0%E7%A7%8D%E6%96%B9%E6%B3%95%E3%80%82%E6%9C%80%E7%AE%80%E5%8D%95%E7%9A%84%E4%B8%80%E7%A7%8D%E6%B6%89%E5%8F%8A%E7%94%B2%E5%9F%BA%E7%9A%84%E8%87%AA%E7%94%B1%E5%9F%BA%E5%8D%A4%E4%BB%A3%E5%92%8C%E5%90%8E%E7%BB%AD%E7%9A%84%E5%88%86%E7%A6%BB%E4%BB%A5%E5%8F%8A%E4%BA%8C%E5%8D%A4%E4%BB%A3%E8%A1%8D%E7%94%9F%E7%89%A9%E7%9A%84%E6%B0%B4%E8%A7%A3%5B%E5%8F%8D%E5%BA%94%289.13%29%5D%EF%BC%9A.jpg "将苄基中心氧化到比可达到的最高氧化态的较低级的水平则显得更为困难。例如,反应ArCH3 ArCHO和反应ArCH2R ArCH(OH)R都是困难的,因为生成的产物比反应物更容易被发生氧化。已经提出了把甲基控制氧化成醛的几种方法。最简单的一种涉及甲基的自由基卤代和后续的分离以及二卤代衍生物的水解[反应(9.13)]:")
133
Example: 选择性褪黑激素受体激动剂:雷美替胺(Ramelteon) 中间体的合成。 日本武田制药株式会社(2005美国首次上市)。
治疗失眠症。 benzofuran-5-carbaldehyde

134
Étard reaction 第二种方法就是利用铬(Ⅵ)试剂在某种条件下确保在最后完成时仅仅产生醛基。在这些氧化反应中最为人们所熟知的是埃塔尔反应(Étard reaction),在惰性溶剂中(CCl4或CS2)进行,氧化剂是铬酰氯,CrO2Cl2,而二乙酸二氧化铬,CrO2(OCOCH3)2:[由氧化铬(VI)、乙酸酐和硫酸就地反应制备]也可成功应用于此目的。在埃塔尔反应中(9.14a),中间体是铬酰氯和甲苯衍生物2:1的加成物,结构可能为8,而在二乙酸二氧化铬氧化中。最初主要产物是二乙酸酯9[反应(9.14b)]:
试剂在某种条件下确保在最后完成时仅仅产生醛基。在这些氧化反应中最为人们所熟知的是埃塔尔反应(Étard reaction),在惰性溶剂中(CCl4或CS2)进行,氧化剂是铬酰氯,CrO2Cl2,而二乙酸二氧化铬,CrO2(OCOCH3)2:[由氧化铬(VI)、乙酸酐和硫酸就地反应制备]也可成功应用于此目的。在埃塔尔反应中(9.14a),中间体是铬酰氯和甲苯衍生物2:1的加成物,结构可能为8,而在二乙酸二氧化铬氧化中。最初主要产物是二乙酸酯9[反应(9.14b)]:")
135
这两种反应并非在所有的例子中都会得到好的产率,但每一类都有一些具有合成价值例子,如:

136
如反应(9.13)所述的一样,要将苄基中心氧化生成醇,可以通过一卤化反应随继发生水解而得以完成。使用四乙酸铅直接氧化到醇的水平也是可能的(参见烯丙位的氧化,第9.2.2节),如:
所述的一样,要将苄基中心氧化生成醇,可以通过一卤化反应随继发生水解而得以完成。使用四乙酸铅直接氧化到醇的水平也是可能的(参见烯丙位的氧化,第9.2.2节),如:")
137
苄基化合物自动氧化,工业上是重要的,但作为“实验室”方法,则价值较小。本节中的许多反应机理尚未完全确定,但大多数情况下,最初一步反应很可能是从苄基碳原子上抽取一个氢自由基或氢负离子。
如果苄基中心失去一个质子很容易,即如果苄基是一个潜在的碳负离子源,它便可与亚硝基芳烃化合物发生缩合,而生成的缩苯胺接着发生水解生成羰基化合物和芳香胺类化合物(参见第6.3.3节),例如:
,例如:")
138
对于苄基碳负离子亚硝化反应的同时还可以导致其发生氧化反应,例如:

139
而脱质子化的一步反应,即10→11,是2-甲基吡啶氧化成2-吡啶基甲醇的关键:将化合物11转化成最后产物可用类似于科普重排形式表示(第7.4.3节):但在化合物11中更有可能是涉及到N-O键断裂而生成离子对或自由基对:
:但在化合物11中更有可能是涉及到N-O键断裂而生成离子对或自由基对:")
140
9.2.4 烷烃、烷基和烯烃的脱氢反应 烷烃和烷基:由相应的二氢化合物脱氢形成烯烃,不能被认为是一个一般反应;只有当双键完全区域专一地引入时,该反应才能成功得以进行,而只有当反应物具有所需结构特征或官能团时这种情况才有可能。尽管如此,在第9.1.1节中所略述的三种脱氢反应是众所周知的,而这些反应也是广泛地应用于引入碳-碳双键的方法。 i)正如(第9.1.1节)已经提到过的那样,在钯或铂存在下的催化脱氢所形成的双键在得以构成芳香体系时,这种脱氢反应才最为成功,如,
正如(第9.1.1节)已经提到过的那样,在钯或铂存在下的催化脱氢所形成的双键在得以构成芳香体系时,这种脱氢反应才最为成功,如,")
141
用于构成稠合芳香体系

142
ii). 在氢原子离子型消除反应中,失去氢负离子通常是反应的第一步[反应(9
. 在氢原子离子型消除反应中,失去氢负离子通常是反应的第一步[反应(9")
144
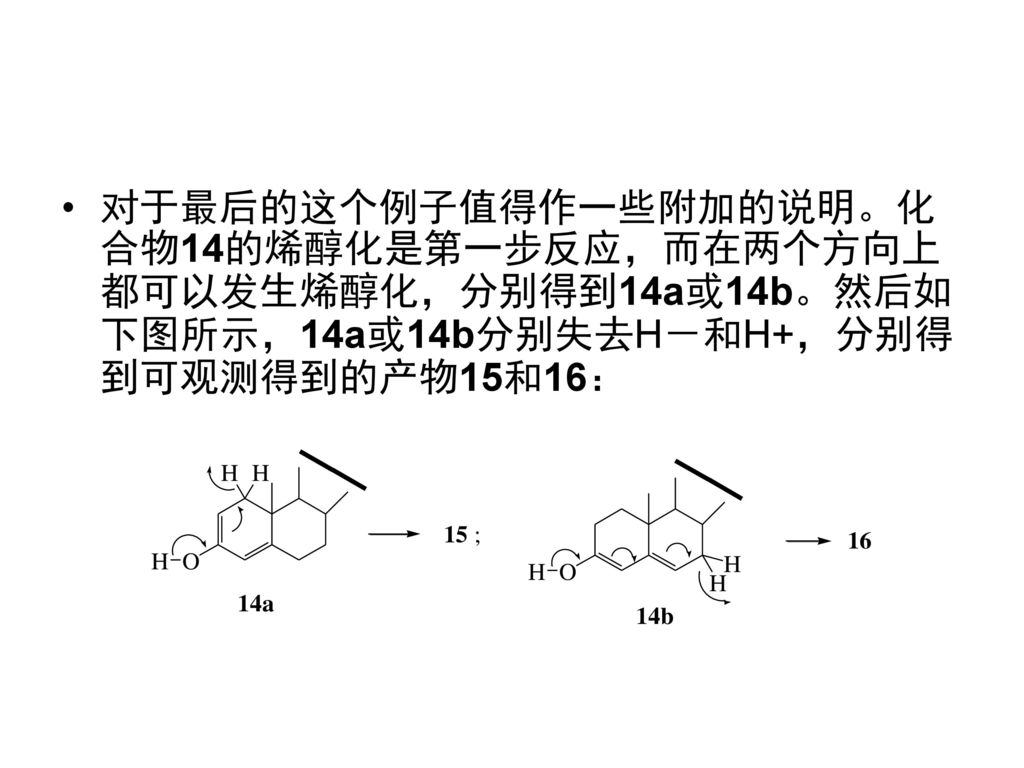
对于最后的这个例子值得作一些附加的说明。化合物14的烯醇化是第一步反应,而在两个方向上都可以发生烯醇化,分别得到14a或14b。然后如下图所示,14a或14b分别失去H-和H+,分别得到可观测得到的产物15和16:

145
iii) 取代-消除程序 最简单的是先卤化(通常是溴化):然后消除卤化氢,如:
在上面每一例中,还应注意溴化和脱溴化氢步骤将会导致生成单一产物, 而该法的成功实施使之可局限用于那些反应时需要遵守区域专一性的情况。

146
烯烃 溴化反应,然后发生脱溴化氢是将烯烃转化为炔烃的一种通用方法,如:

147
9.2.5 对烯烃的氧化加成反应 在这一节中,我们将考虑两种类型的加成反应:通过把一个氧原子加到双键上而形成环氧乙烷类化合物(环氧化物)以及1, 2-二醇化合物的形成,后者实际上是在双键的两端各加上一个羟基。
以及1, 2-二醇化合物的形成,后者实际上是在双键的两端各加上一个羟基。")
148
环氧乙烷的形成(环氧化) 环氧乙烷类化合物(即环氧化物)是由烯烃和过氧酸直接反应而形成的[反应(9.16):参见赛克斯,第190页],在产物中仍然保持着烯烃的立体化学:
![环氧乙烷的形成(环氧化) 环氧乙烷类化合物(即环氧化物)是由烯烃和过氧酸直接反应而形成的[反应(9.16):参见赛克斯,第190页],在产物中仍然保持着烯烃的立体化学:](http://slidesplayer.com/slide/11130619/59/images/148/%E7%8E%AF%E6%B0%A7%E4%B9%99%E7%83%B7%E7%9A%84%E5%BD%A2%E6%88%90%28%E7%8E%AF%E6%B0%A7%E5%8C%96%29+%E7%8E%AF%E6%B0%A7%E4%B9%99%E7%83%B7%E7%B1%BB%E5%8C%96%E5%90%88%E7%89%A9%28%E5%8D%B3%E7%8E%AF%E6%B0%A7%E5%8C%96%E7%89%A9%29%E6%98%AF%E7%94%B1%E7%83%AF%E7%83%83%E5%92%8C%E8%BF%87%E6%B0%A7%E9%85%B8%E7%9B%B4%E6%8E%A5%E5%8F%8D%E5%BA%94%E8%80%8C%E5%BD%A2%E6%88%90%E7%9A%84%5B%E5%8F%8D%E5%BA%94%289.16%29%EF%BC%9A%E5%8F%82%E8%A7%81%E8%B5%9B%E5%85%8B%E6%96%AF%EF%BC%8C%E7%AC%AC190%E9%A1%B5%5D%EF%BC%8C%E5%9C%A8%E4%BA%A7%E7%89%A9%E4%B8%AD%E4%BB%8D%E7%84%B6%E4%BF%9D%E6%8C%81%E7%9D%80%E7%83%AF%E7%83%83%E7%9A%84%E7%AB%8B%E4%BD%93%E5%8C%96%E5%AD%A6%EF%BC%9A.jpg "环氧乙烷的形成(环氧化) 环氧乙烷类化合物(即环氧化物)是由烯烃和过氧酸直接反应而形成的[反应(9.16):参见赛克斯,第190页],在产物中仍然保持着烯烃的立体化学:")
149
过硼酸钠和过碳酸钠 过氧酸常常能够就地由过氧化氢和羧酸衍生物作用而产生。浓缩的过氧化氢溶液的处理上存在着严重的问题,然而,过硼酸钠(SPB, 17)和过碳酸钠(SPC, 18)作为固体化合物则为该试剂提供了另外的便利选择,因为它们通过与合适的羧酸或者酸酐反应可以直接得到过氧酸。 间-氯过氧苯甲酸(m-ClC6H4COOOH;常常被称为是MCPBA)也是一个相对稳定的固体。
和过碳酸钠(SPC, 18)作为固体化合物则为该试剂提供了另外的便利选择,因为它们通过与合适的羧酸或者酸酐反应可以直接得到过氧酸。 间-氯过氧苯甲酸(m-ClC6H4COOOH;常常被称为是MCPBA)也是一个相对稳定的固体。")
150
过氧化作用例子 过硼酸钠

151
与一个-M基团共轭的烯烃通过与碱性的过氧化氢反应而被环氧化:这个反应涉及HO2 - 的一种类似迈克加成反应继而失去OH - [反应(9
与一个-M基团共轭的烯烃通过与碱性的过氧化氢反应而被环氧化:这个反应涉及HO2 - 的一种类似迈克加成反应继而失去OH - [反应(9.17)]。应当注意,在这种情况下,产物不必保持原来烯烃的立体化学,因为在其中间体中有可能围绕其C2-C3 键进行自由旋转:
]。应当注意,在这种情况下,产物不必保持原来烯烃的立体化学,因为在其中间体中有可能围绕其C2-C3 键进行自由旋转:")
152
,2-二醇的形成(羟基化) 将烯烃转化生成1,2-二醇类化合物的常用方法一般有三种;这些方法之间在一定程度上将会互相补充,因而全部都值得考虑和讨论。第一种方法经由环氧乙烷的过程,将会导致生成反式加成物[反应(9.18)]。
![,2-二醇的形成(羟基化) 将烯烃转化生成1,2-二醇类化合物的常用方法一般有三种;这些方法之间在一定程度上将会互相补充,因而全部都值得考虑和讨论。第一种方法经由环氧乙烷的过程,将会导致生成反式加成物[反应(9.18)]。](http://slidesplayer.com/slide/11130619/59/images/152/%2C2-%E4%BA%8C%E9%86%87%E7%9A%84%E5%BD%A2%E6%88%90%28%E7%BE%9F%E5%9F%BA%E5%8C%96%29+%E5%B0%86%E7%83%AF%E7%83%83%E8%BD%AC%E5%8C%96%E7%94%9F%E6%88%901%2C2-%E4%BA%8C%E9%86%87%E7%B1%BB%E5%8C%96%E5%90%88%E7%89%A9%E7%9A%84%E5%B8%B8%E7%94%A8%E6%96%B9%E6%B3%95%E4%B8%80%E8%88%AC%E6%9C%89%E4%B8%89%E7%A7%8D%EF%BC%9B%E8%BF%99%E4%BA%9B%E6%96%B9%E6%B3%95%E4%B9%8B%E9%97%B4%E5%9C%A8%E4%B8%80%E5%AE%9A%E7%A8%8B%E5%BA%A6%E4%B8%8A%E5%B0%86%E4%BC%9A%E4%BA%92%E7%9B%B8%E8%A1%A5%E5%85%85%EF%BC%8C%E5%9B%A0%E8%80%8C%E5%85%A8%E9%83%A8%E9%83%BD%E5%80%BC%E5%BE%97%E8%80%83%E8%99%91%E5%92%8C%E8%AE%A8%E8%AE%BA%E3%80%82%E7%AC%AC%E4%B8%80%E7%A7%8D%E6%96%B9%E6%B3%95%E7%BB%8F%E7%94%B1%E7%8E%AF%E6%B0%A7%E4%B9%99%E7%83%B7%E7%9A%84%E8%BF%87%E7%A8%8B%EF%BC%8C%E5%B0%86%E4%BC%9A%E5%AF%BC%E8%87%B4%E7%94%9F%E6%88%90%E5%8F%8D%E5%BC%8F%E5%8A%A0%E6%88%90%E7%89%A9%5B%E5%8F%8D%E5%BA%94%289.18%29%5D%E3%80%82.jpg ",2-二醇的形成(羟基化) 将烯烃转化生成1,2-二醇类化合物的常用方法一般有三种;这些方法之间在一定程度上将会互相补充,因而全部都值得考虑和讨论。第一种方法经由环氧乙烷的过程,将会导致生成反式加成物[反应(9.18)]。")
153
该方法取决于酸RCOOH的强度(即取决于它使环氧乙烷的氧原子质子化的能力)。甲酸和三氟乙酸的强度足以影响到开环,因而其过氧化物的衍生物则常用于羟基化。像这样的例子列举如下:
。甲酸和三氟乙酸的强度足以影响到开环,因而其过氧化物的衍生物则常用于羟基化。像这样的例子列举如下:")
154
第二种方法涉及到环酯形成,是通过烯烃和高锰酸钾或四氧化锇(VIII)反应[反应(9.19)],接着发生水解将导致产生顺式构型加成物。
![第二种方法涉及到环酯形成,是通过烯烃和高锰酸钾或四氧化锇(VIII)反应[反应(9.19)],接着发生水解将导致产生顺式构型加成物。](http://slidesplayer.com/slide/11130619/59/images/154/%E7%AC%AC%E4%BA%8C%E7%A7%8D%E6%96%B9%E6%B3%95%E6%B6%89%E5%8F%8A%E5%88%B0%E7%8E%AF%E9%85%AF%E5%BD%A2%E6%88%90%EF%BC%8C%E6%98%AF%E9%80%9A%E8%BF%87%E7%83%AF%E7%83%83%E5%92%8C%E9%AB%98%E9%94%B0%E9%85%B8%E9%92%BE%E6%88%96%E5%9B%9B%E6%B0%A7%E5%8C%96%E9%94%87%28VIII%29%E5%8F%8D%E5%BA%94%5B%E5%8F%8D%E5%BA%94%289.19%29%5D%EF%BC%8C%E6%8E%A5%E7%9D%80%E5%8F%91%E7%94%9F%E6%B0%B4%E8%A7%A3%E5%B0%86%E5%AF%BC%E8%87%B4%E4%BA%A7%E7%94%9F%E9%A1%BA%E5%BC%8F%E6%9E%84%E5%9E%8B%E5%8A%A0%E6%88%90%E7%89%A9%E3%80%82.jpg "第二种方法涉及到环酯形成,是通过烯烃和高锰酸钾或四氧化锇(VIII)反应[反应(9.19)],接着发生水解将导致产生顺式构型加成物。")
155
然而,四氧化锇(VIII)价格不仅要比高锰酸钾贵几百多倍,且有剧毒。因此,它仅可用于那些小规模反应,其成本相对较低而且产率则必须很高。高锰酸钾虽价格便宜,但可引起二醇进一步氧化(参见第9.3.2节),还可使复杂分子中的其他官能团也发生氧化;因此,采用高锰酸盐进行羟基化反应产率并不总是很高。实例包括:
价格不仅要比高锰酸钾贵几百多倍,且有剧毒。因此,它仅可用于那些小规模反应,其成本相对较低而且产率则必须很高。高锰酸钾虽价格便宜,但可引起二醇进一步氧化(参见第9.3.2节),还可使复杂分子中的其他官能团也发生氧化;因此,采用高锰酸盐进行羟基化反应产率并不总是很高。实例包括:")
156
上述例子中需注意:在可能的二醇产物中,仅分离出一种顺式构型;这是由于高锰酸盐是从空间位阻较小的一边进攻烯烃的缘故。

157
Prévost Reaction 第三种羟基化的方法是普雷沃(Prévost)反应,在这个反应中,烯烃与碘和银盐(通常是苯甲酸银盐或乙酸银盐)共热,根据反应条件,这个方法可导致对双键进行顺式或反式加成[反应(9.20)]: 碘代酯19
![Prévost Reaction 第三种羟基化的方法是普雷沃(Prévost)反应,在这个反应中,烯烃与碘和银盐(通常是苯甲酸银盐或乙酸银盐)共热,根据反应条件,这个方法可导致对双键进行顺式或反式加成[反应(9.20)]:](http://slidesplayer.com/slide/11130619/59/images/157/Pr%C3%A9vost+Reaction+%E7%AC%AC%E4%B8%89%E7%A7%8D%E7%BE%9F%E5%9F%BA%E5%8C%96%E7%9A%84%E6%96%B9%E6%B3%95%E6%98%AF%E6%99%AE%E9%9B%B7%E6%B2%83%28Pr%C3%A9vost%29%E5%8F%8D%E5%BA%94%EF%BC%8C%E5%9C%A8%E8%BF%99%E4%B8%AA%E5%8F%8D%E5%BA%94%E4%B8%AD%EF%BC%8C%E7%83%AF%E7%83%83%E4%B8%8E%E7%A2%98%E5%92%8C%E9%93%B6%E7%9B%90%28%E9%80%9A%E5%B8%B8%E6%98%AF%E8%8B%AF%E7%94%B2%E9%85%B8%E9%93%B6%E7%9B%90%E6%88%96%E4%B9%99%E9%85%B8%E9%93%B6%E7%9B%90%29%E5%85%B1%E7%83%AD%EF%BC%8C%E6%A0%B9%E6%8D%AE%E5%8F%8D%E5%BA%94%E6%9D%A1%E4%BB%B6%EF%BC%8C%E8%BF%99%E4%B8%AA%E6%96%B9%E6%B3%95%E5%8F%AF%E5%AF%BC%E8%87%B4%E5%AF%B9%E5%8F%8C%E9%94%AE%E8%BF%9B%E8%A1%8C%E9%A1%BA%E5%BC%8F%E6%88%96%E5%8F%8D%E5%BC%8F%E5%8A%A0%E6%88%90%5B%E5%8F%8D%E5%BA%94%289.20%29%5D%EF%BC%9A.jpg "碘代酯19.")
158
Examples 对烯烃的最初反式加成将会得到碘代酯19,在没有任何其他亲核试剂存在下,
应当注意,由于邻近基团的参与将会使得所得到的二酯(由之可以得到二醇)保持反式的立体化学。然而,在水存在下,碘代酯可能会以不同的方式进行水解[反应(9.20b)],从而得到顺式二醇。
保持反式的立体化学。然而,在水存在下,碘代酯可能会以不同的方式进行水解[反应(9.20b)],从而得到顺式二醇。")
159
因此,在无水的条件下,通过普雷沃反应得到产物和经由过氧酸反应所得产物相同,都是二醇的结构,例如:

160
Woodward modification
普雷沃反应只不过是对其他令人满意的羟基化方法的一种价格昂贵的替代方法。 然而,就将对酸敏感化合物的反式羟基化而言,与过氧酸方法相比具有明显优点。

161
伍德华改良在用于从一种烯烃生成两种顺式二醇时,则是一种重要的方法;高锰酸盐氧化则产生位阻较小的二醇(参见上面),伍德华方法导致生成较少位阻的碘正离子而由此得位阻较大的二醇,如:
,伍德华方法导致生成较少位阻的碘正离子而由此得位阻较大的二醇,如:")
162
硝酸铈铵(CAN) CAN:(NH4)2Ce(NO3)6是一种单电子氧化试剂。在温和条件下会发生各种选择性氧化反应。在烯烃例子中,产物结构可能取决于溶剂种类和外加亲电试剂。
 CAN:(NH4)2Ce(NO3)6是一种单电子氧化试剂。在温和条件下会发生各种选择性氧化反应。在烯烃例子中,产物结构可能取决于溶剂种类和外加亲电试剂。")
163
9.2.6 烯烃的氧化断裂反应 这种反应类型在合成上的价值比它在降解法测定结构方面的价值要小得多。碳-碳双键的氧化断裂常导致生成醛、酮或羧酸,且很少能看到烯烃能够作为一种最方便的来源来合成其中任何一种化合物。但是在第7.4.2节中我们已注意到了氧化断裂作为开环方法的价值,而在多步骤合成中,烯烃偶尔也能作为潜在的羰基官能团。有两种基本方法可用于引起这种断裂。

164
第一种方法涉及到首先进行羟基化,然后发生二醇的氧化反应(使用高锰酸钾,四乙酸铅或过氧酸盐,参见第9.3.2节)。
第二种方法涉及烯烃的臭氧化反应[反应(9.21)],即涉及臭氧的1,3-偶极环加成,周环开环(“逆-环加成”)及再一次1,3-偶极环加成的一系列反应:
],即涉及臭氧的1,3-偶极环加成,周环开环( 逆-环加成 )及再一次1,3-偶极环加成的一系列反应:")
165
反应主要产物臭氧化物(21)没被分离出来,但可直接转化成所需羰基化合物。经还原处理后(例如,用锌和乙酸或络合金属氢化物,或三价磷试剂)产生醛或酮(尽管还原试剂过量可能会与这些化合物作用:如,使用氢化锂铝得醇): 或

166
氧化处理 氧化处理般牵涉到过氧酸,而且在其第一步反应中,还可能包含有水解过程。在这样条件下,产物是羧酸或酮:

167
9.2.7 炔烃的氧化反应 将碳-碳三键进行氧化的合成过程比对烯烃的相映氧化更少被人们所采用。我们现在已知的一些相关例子是通过羟基化的方法来形成1,2-二酮: 然而在许多情况下,该反应得到的是复杂混合产物, 因此,炔烃羟基化并不是制备二酮的一种通用方法。 不过,作为一种极其重要常用方法是l-炔烃的氧化偶合。

168
2.3 醇及其衍生物的氧化反应 9.3.1 醛或酮的形成(脱氢)
2.3 醇及其衍生物的氧化反应 9.3.1 醛或酮的形成(脱氢) 大多数学习有机化学的学生在早期已经懂得了由伯醇氧化的早期可以得到醛,然后进一步氧化可以得到羧酸,仲醇氧化则可以得到酮,而叔醇则是耐氧化的,除非反应条件极为激烈时发生碳-碳键的断裂。通常,被编写入基础教材的氧化反应是完全没有选择性的,使用的试剂像热的高锰酸钾或热的铬酸。但是,为了获得把醇选择性氧化成羰基化合物的方法,人们已经进行了大量的相关研究工作。 将醇转化成羰基化合物的反应形式上是一种脱氢作用,在第9.1.1节中所概述的三种通用的方法均可以运用于此。
 大多数学习有机化学的学生在早期已经懂得了由伯醇氧化的早期可以得到醛,然后进一步氧化可以得到羧酸,仲醇氧化则可以得到酮,而叔醇则是耐氧化的,除非反应条件极为激烈时发生碳-碳键的断裂。通常,被编写入基础教材的氧化反应是完全没有选择性的,使用的试剂像热的高锰酸钾或热的铬酸。但是,为了获得把醇选择性氧化成羰基化合物的方法,人们已经进行了大量的相关研究工作。 将醇转化成羰基化合物的反应形式上是一种脱氢作用,在第9.1.1节中所概述的三种通用的方法均可以运用于此。")
169
9.3.1 醛或酮的形成(脱氢) 取代-消除方法 取代-消除反应至少在实验室规模上是极为普通。为把仲醇氧化成酮,铬(VI)氧化剂是最常用的试剂。反应经由铬酸酯进行[如反应(9.2.2)]:
氧化剂是最常用的试剂。反应经由铬酸酯进行[如反应(9.2.2)]: .")
170
经由第一步所产生的四价Cr(IV)衍生物不是最终产物。一个更复杂的氧化还原步骤将最后导致生成三价铬(III)盐,而该反应总化学计量转化式如下:
或: 或:

171
我们现已熟悉对于这种氧化作用的许多变通方法。如果醇分子中不含有其他可被氧化的官能团,并且对酸不敏感时,则采用溶于硫酸水溶液或乙酸水溶液的铬酸进行氧化最方便,如:

172
使用琼斯试剂(Jones’ reagent)可以将含有双键或叁键的醇进行选择性的氧化,该试剂是由氧化铬(VI)和硫酸以适当的化学计量按比例组成的水溶液;实际上,在室温或低于室温时,该试剂可以采用来滴定溶解在丙酮中的醇。在这些条件下,醇基则被选择性地氧化,例如:
可以将含有双键或叁键的醇进行选择性的氧化,该试剂是由氧化铬(VI)和硫酸以适当的化学计量按比例组成的水溶液;实际上,在室温或低于室温时,该试剂可以采用来滴定溶解在丙酮中的醇。在这些条件下,醇基则被选择性地氧化,例如:")
173
假如遇到对酸敏感性问题,则可以选择在吡啶中的氧化铬(VI)作为氧化剂。换句话说,可将氧化铬-吡啶络合物分离出来,并在另一种有机溶剂中使用,比如二氯甲烷。如:
作为氧化剂。换句话说,可将氧化铬-吡啶络合物分离出来,并在另一种有机溶剂中使用,比如二氯甲烷。如:")
174
用于在有机溶剂中氧化的铬(VI)试剂包括吡啶氯化铬酸盐[(C5H5N+HCrO3Cl-,PCC),由氧化铬(VI),盐酸和吡啶反应而制得]和吡啶重铬酸盐[(C5H5N+H)2Cr2O72-,PDC],它是由氧化铬(VI),吡啶和水反应而制得。例如:
![用于在有机溶剂中氧化的铬(VI)试剂包括吡啶氯化铬酸盐[(C5H5N+HCrO3Cl-,PCC),由氧化铬(VI),盐酸和吡啶反应而制得]和吡啶重铬酸盐[(C5H5N+H)2Cr2O72-,PDC],它是由氧化铬(VI),吡啶和水反应而制得。例如:](http://slidesplayer.com/slide/11130619/59/images/174/%E7%94%A8%E4%BA%8E%E5%9C%A8%E6%9C%89%E6%9C%BA%E6%BA%B6%E5%89%82%E4%B8%AD%E6%B0%A7%E5%8C%96%E7%9A%84%E9%93%AC%28VI%29%E8%AF%95%E5%89%82%E5%8C%85%E6%8B%AC%E5%90%A1%E5%95%B6%E6%B0%AF%E5%8C%96%E9%93%AC%E9%85%B8%E7%9B%90%5B%28C5H5N%2BHCrO3Cl-%EF%BC%8CPCC%29%EF%BC%8C%E7%94%B1%E6%B0%A7%E5%8C%96%E9%93%AC%28VI%29%EF%BC%8C%E7%9B%90%E9%85%B8%E5%92%8C%E5%90%A1%E5%95%B6%E5%8F%8D%E5%BA%94%E8%80%8C%E5%88%B6%E5%BE%97%5D%E5%92%8C%E5%90%A1%E5%95%B6%E9%87%8D%E9%93%AC%E9%85%B8%E7%9B%90%5B%28C5H5N%2BH%292Cr2O72-%EF%BC%8CPDC%5D%EF%BC%8C%E5%AE%83%E6%98%AF%E7%94%B1%E6%B0%A7%E5%8C%96%E9%93%AC%28VI%29%EF%BC%8C%E5%90%A1%E5%95%B6%E5%92%8C%E6%B0%B4%E5%8F%8D%E5%BA%94%E8%80%8C%E5%88%B6%E5%BE%97%E3%80%82%E4%BE%8B%E5%A6%82%EF%BC%9A.jpg "用于在有机溶剂中氧化的铬(VI)试剂包括吡啶氯化铬酸盐[(C5H5N+HCrO3Cl-,PCC),由氧化铬(VI),盐酸和吡啶反应而制得]和吡啶重铬酸盐[(C5H5N+H)2Cr2O72-,PDC],它是由氧化铬(VI),吡啶和水反应而制得。例如:")
175
对于含有在酸性条件下敏感的官能团的醇,PDC是优先选择的试剂。用于这类反应其他一些氧化剂还有2,2’-双吡啶基鎓氯铬酸盐(22)和吡嗪鎓氯铬酸盐(23)。采用22可以很明显地简化后处理步骤(所有含铬的副产物都是水溶性的),而对于23也具有类似的优点(产物可以通过简单的萃取和色谱的方法进行分离)。
和吡嗪鎓氯铬酸盐(23)。采用22可以很明显地简化后处理步骤(所有含铬的副产物都是水溶性的),而对于23也具有类似的优点(产物可以通过简单的萃取和色谱的方法进行分离)。")
176
对于将伯醇氧化成醛,必须小心地控制反应条件以防止氧化过度而产生羧酸。对于这种氧化来说,选择铬(VI)试剂仍然是一种普遍使用的方法。制备低级脂肪醛的经典方法,就是利用其相对低的沸点,当产物一旦形成就可以从氧化性的溶液中被蒸馏出来:例如:
试剂仍然是一种普遍使用的方法。制备低级脂肪醛的经典方法,就是利用其相对低的沸点,当产物一旦形成就可以从氧化性的溶液中被蒸馏出来:例如:")
177
对于不易挥发的醛,在某些情况下,通过严格地控制反应时间和温度也可能获得良好的产率,例如:

178
????? 这种类型的氧化反应,选择PCC和PDC试剂看来似乎是一种普遍适用的方法,例如: 同样地,

179
在其他情况下(特别是涉及到烯丙位或苄基位氧化时,使用CrO3/吡啶试剂将是令人满意的,例如:

180
Swern氧化反应

181
D. Swern 发现背景 76:DMSO+TFAA,-50℃,DCM得锍盐,和伯醇和仲醇快速发生反应得烷氧基二甲基锍三氟乙酸盐衍生物,加入三乙胺,可以高收率获得相应的醛和酮类衍生物。 78:草酰氯可替代TFAA用作DMSO的活化试剂。

182
反应的八大特点 1) 当没有使用溶剂参加反应时,DMSO和TFAA或草酰氯会剧烈反应(爆炸!),所以操作反应时应该多加小心;
2) 最常用的溶剂是DCM; 3) 当用TFFA时,反应最初的中间体在-30℃以上不稳定,并会通过Pummerer重排反应而产生一种副产物; 4) 如果利用草酰氯,反应最初的中间体在-60℃以上不稳定,所以该氧化反应常需要控制在-78℃进行; 5) 典型的操作步骤是首先用DMSO和TFAA或草酰氯在低温下进行反应,然后再慢慢加入醇,接着加入的是叔胺; 6) 要加速烷氧基锍鎓盐的分解则必须添加叔胺(如,DIPA,TEA); 7) 氧化反应的效率不受反应底物的立体位阻效应的影响; 8) 使用TFAA可能会生成副产物三氟醋酸酯,而若使用草酰氯时,该副反应则极端少见。
 最常用的溶剂是DCM; 3) 当用TFFA时,反应最初的中间体在-30℃以上不稳定,并会通过Pummerer重排反应而产生一种副产物; 4) 如果利用草酰氯,反应最初的中间体在-60℃以上不稳定,所以该氧化反应常需要控制在-78℃进行; 5) 典型的操作步骤是首先用DMSO和TFAA或草酰氯在低温下进行反应,然后再慢慢加入醇,接着加入的是叔胺; 6) 要加速烷氧基锍鎓盐的分解则必须添加叔胺(如,DIPA,TEA); 7) 氧化反应的效率不受反应底物的立体位阻效应的影响; 8) 使用TFAA可能会生成副产物三氟醋酸酯,而若使用草酰氯时,该副反应则极端少见。")
183
反应机理:DMSO/TFAA

184
反应机理:DMSO/Oxalyl chloride

185
应用: Dolabellane二萜首次全合成
海洋产物Dolabellane二萜的(+)脱氧新Dolabellane的首次全合成是在D.R.Wiliiams的实验室中完成的。17在合成步骤中的最后一步反应,是需要将1,2-二醇的仲醇官能团的氧化得到相应的α-羟基酮。已知1,2-二醇是在大多数的氧化反应条件下都不稳定,常常会观察到乙二醇的裂解。事实上,当尝试了Dess-Martin和Ley氧化反应之时,底物的碳-碳键发生了断裂。然而,在Swern氧化反应的条件下,所希望的α-羟基酮的分离产率是65%。有趣的是,采用的反应底物是四种不可分离的非对映异构体的二醇结构(是通过McMury反应而获得的),却得到了两种容易分离的酮类产物,其中之一是天然产物。
脱氧新Dolabellane的首次全合成是在D.R.Wiliiams的实验室中完成的。17在合成步骤中的最后一步反应,是需要将1,2-二醇的仲醇官能团的氧化得到相应的α-羟基酮。已知1,2-二醇是在大多数的氧化反应条件下都不稳定,常常会观察到乙二醇的裂解。事实上,当尝试了Dess-Martin和Ley氧化反应之时,底物的碳-碳键发生了断裂。然而,在Swern氧化反应的条件下,所希望的α-羟基酮的分离产率是65%。有趣的是,采用的反应底物是四种不可分离的非对映异构体的二醇结构(是通过McMury反应而获得的),却得到了两种容易分离的酮类产物,其中之一是天然产物。")
186
生物碱Ircinal A的合成 S.F.Matin和他的合作者们在对Ircinal A和相关的Manzamine生物碱类化合物进行合成的过程中,采用了双重Swern氧化反应。高级三环二醇中间体首先在-78℃下经历Swern氧化反应的条件,得到相应的二醛,产率极佳。接着在下一步反应中,将此二醛处于过量的Wittig试剂中,在无盐的条件下形成两个端基烯烃。

187
(+)-Asteltoxin的汇聚式全合成
具有有丝分裂毒性的(+)-Asteltoxin的汇聚式全合成是由J.K.Cha等人完成的。其中两个主要片段的偶合是由双(四氢呋喃)甲醛和α-吡喃酮磷酸酯经Horner-Wadsworth-Emmons(HWE)烯烃化反应而完成的。双(四氢呋喃)甲醛是由双(四氢呋喃)伯醇经过Swern氧化反应得到的。有趣的是,在该氧化反应的条件下,没有α-立体中心的差向异构化,但是在HWE烯烃化反应过程中却形成了少量的C8差向异构体。 Horner-Wadsworth-Emmons: HWE烯烃化反应: L.Horner利用烷基二苯基磷氧化物中的碳负离子和醛酮反应来制备烯烃
-Asteltoxin的汇聚式全合成是由J.K.Cha等人完成的。其中两个主要片段的偶合是由双(四氢呋喃)甲醛和α-吡喃酮磷酸酯经Horner-Wadsworth-Emmons(HWE)烯烃化反应而完成的。双(四氢呋喃)甲醛是由双(四氢呋喃)伯醇经过Swern氧化反应得到的。有趣的是,在该氧化反应的条件下,没有α-立体中心的差向异构化,但是在HWE烯烃化反应过程中却形成了少量的C8差向异构体。 Horner-Wadsworth-Emmons: HWE烯烃化反应: L.Horner利用烷基二苯基磷氧化物中的碳负离子和醛酮反应来制备烯烃.")
188
Example:鲁比前列酮中间体的合成 双环伯醇11-2-20在低温条件下经Swern氧化反应得醇氧化得醛。
鲁比前列酮的合成路线三的关键步骤

189
鲁比前列酮中间体的合成

190
氢化物转移法

191
其他方法

192
,2-二醇的氧化断裂

193
2.4 芳香体系的氧化反应 9.4.1 芳香烃的氧化反应

194
2.4.1芳香烃的氧化反应

195
2.5 醛和酮的氧化反应 2.5.1 氧化成羧酸

196
2.5.2 氧化成1,2-二羰基化合物

197
2.5.3 氧化成酯 拜耳-维利格(Baeyer-Villiger) 达金(Dakin)反应
 达金(Dakin)反应")
198
2.6 含氮官能团的氧化反应 9.6.1 N-氧化的化合物的形成

199
2.6.2涉及含氮官能团的脱氢作用

200
2.6.3 硝基烷烃的氧化反应

201
2.7 含硫官能团的氧化反应 2.7.1 硫醇

202
2.7.2 硫化物 硫醚和其衍生物,亚砜、砜、磺化物,等。

203
治疗胃溃疡、十二指肠溃疡、消化性食管炎及胃炎。
Example:硫醚氧化为亚砜 质子泵抑制剂: 埃索美拉唑钠 (Esomeprazole Sodium) 的合成的最后一步氧化反应。 瑞典Astra Zeneca 公司: Nexium ® (耐信 ®) 、美国FDA批准上市(1999). 治疗胃溃疡、十二指肠溃疡、消化性食管炎及胃炎。 以4-甲氧基-3,5-二甲基-2-羟甲基吡啶为原料,经氯化后与5-甲氧基-2-巯基苯并咪唑反应合成奥美拉唑硫醚物,奥美拉唑硫醚物在以( 1S,2 S) -( - ) -1,2 -环己二胺-D-酒石酸盐为手性配体、环烷酸钴为催化剂、过氧化钠为氧化剂的条件下进行不对称氧化合成高光学纯度埃索美拉唑钠。 References: 宋伟国; 张祥敏; 张晓攀; 徐文芳. 中国药物化学杂志 2013, 23(4), 286. 硫醚的立体选择性氧化需要手性催化助剂。
 的合成的最后一步氧化反应。 瑞典Astra Zeneca 公司: Nexium ® (耐信 ®) 、美国FDA批准上市(1999). 治疗胃溃疡、十二指肠溃疡、消化性食管炎及胃炎。 以4-甲氧基-3,5-二甲基-2-羟甲基吡啶为原料,经氯化后与5-甲氧基-2-巯基苯并咪唑反应合成奥美拉唑硫醚物,奥美拉唑硫醚物在以( 1S,2 S) -( - ) -1,2. -环己二胺-D-酒石酸盐为手性配体、环烷酸钴为催化剂、过氧化钠为氧化剂的条件下进行不对称氧化合成高光学纯度埃索美拉唑钠。 References: 宋伟国; 张祥敏; 张晓攀; 徐文芳. 中国药物化学杂志 2013, 23(4), 286. 硫醚的立体选择性氧化需要手性催化助剂。")
204
治疗十二指肠溃疡与胃溃疡、反流性食管炎等。
Example: 雷贝拉唑钠的合成 抗胃溃疡药:雷贝拉唑钠 (Rabeprazole sodium )中间体的合成。 日本卫材公司(1998日本上市)。 治疗十二指肠溃疡与胃溃疡、反流性食管炎等。 2-((4-(3-methoxypropoxy)-3-methylpyridin -2-yl)methylsulfinyl)-1H-benzo[d]imidazole 与NaOH成盐后得雷贝拉唑钠。
中间体的合成。 日本卫材公司(1998日本上市)。 治疗十二指肠溃疡与胃溃疡、反流性食管炎等。 2-((4-(3-methoxypropoxy)-3-methylpyridin. -2-yl)methylsulfinyl)-1H-benzo[d]imidazole. 与NaOH成盐后得雷贝拉唑钠。")
205
NSAID:罗非昔布:Ⅱ型环氧化酶(COX-2)抑制剂,用于缓解骨关节炎症状。
MMPP:邻苯二甲酸镁的六水合物:一个廉价、安全、市场可购买的MCPBA的替代品。 MCPBA:间氯过氧苯甲酸

206
罗非昔布( ) 生产方法: 197g4-甲硫基苯甲酮溶于700mL甲醇和350mL二氯甲烷中,在30min内加入881gMMPP,在室温搅拌3h。过滤,滤液用2L饱和碳酸氢钠水溶液和1L饱和食盐水洗。水溶液再用2L二氯甲烷萃取,萃取液和有机层合并,干燥,浓缩,得240g白色固体的4-甲磺酰基苯甲酮。 174g4-甲磺酰基苯甲酮溶于2.5L氯仿,在-5℃下,加入20mg三氯化铝,再加入40mL溴在300mL氯仿中的溶液。反应后,加入1.5L水,分去氯仿,水层用1L乙酸乙酯萃取。萃取液合并,干燥,浓缩。粗品用50/50的乙酸乙酯/己烷重结晶,得210g白色固体的2-溴-1-(4-甲磺酰基苯基)乙酮。 在20℃,27.4g苯乙酸和60g2-溴-1-(4-甲磺酰基苯基)乙酮溶于630mL乙腈,再缓慢加入30.8mL三乙胺。在室温搅拌20min后,用冰浴冷却。缓慢加入60.1mLDBU,搅拌20min至反应完全。用1mol/L盐酸酸化(颜色从深棕色变为黄色),然后加入2.4L冰水混合物,搅拌少许。过滤析出的沉淀,少量水洗,得到64g湿的粗品。将其溶于750mL二氯甲烷,加入300g硅胶。浓缩至略干,剩余物装入硅胶柱的顶部,用10%乙酸乙酯/二氯甲烷层析。展开液浓缩后,得36.6g产物。收率58%。
乙酮。 在20℃,27.4g苯乙酸和60g2-溴-1-(4-甲磺酰基苯基)乙酮溶于630mL乙腈,再缓慢加入30.8mL三乙胺。在室温搅拌20min后,用冰浴冷却。缓慢加入60.1mLDBU,搅拌20min至反应完全。用1mol/L盐酸酸化(颜色从深棕色变为黄色),然后加入2.4L冰水混合物,搅拌少许。过滤析出的沉淀,少量水洗,得到64g湿的粗品。将其溶于750mL二氯甲烷,加入300g硅胶。浓缩至略干,剩余物装入硅胶柱的顶部,用10%乙酸乙酯/二氯甲烷层析。展开液浓缩后,得36.6g产物。收率58%。")
207
Synthesis route of Rofecoxib
环化缩合策略的原始合成法

208
m-CPBA Xingmin Zhang, Anjun Hu,* Chongfeng Pan, Qiwu Zhao, Xianwen Wang, and Jiang Lu,Safer Preparation of m-CPBA/DMF Solution in Pilot Plant, OPRD, 2013,

209
Example:硒醚氧化为砜 Example:硒代农药,除草活性 Oxone 过硫酸氢钾的复盐
刘润辉,李忠,黄青春,宋恭华,含硒化合物的合成及农药活性研究, 农药, Oxone 过硫酸氢钾的复盐

Similar presentations

 3、一些常用的不饱和基团(烯基)>")








基化反应 醛(酮)及酯的缩合反应 生成碳碳双键的反应 成环的反应 其他类型的负碳离子反应>")
是由30个碳原子组成的萜类化合物。(指基本骨架,不包括糖),可认为是由6个异戊二烯缩合而成的。 分类 从结构上分两大类:四环三萜 五环三萜 存在形式:游离形式(苷元) 苷的形式(与糖结合)>")





>")

