Download presentation
1
第六章 离子聚合

2
6.1 引言 离子聚合:活性中心是离子的聚合。 阳离子聚合 根据中心离子电荷性质的不同 阴离子聚合 离子聚合的理论研究开始于五十年代:
1953年,Ziegler在常温低压下制得PE 1956年,Szwarc发现了“活性聚合物” 多数烯烃单体都能进行自由基聚合,但是离子聚合却有极高的选择性。 原因: 离子聚合对阳离子和阴离子的稳定性要求比较严格。

3
带有1,1-二烷基、烷氧基等推电子基的单体才能进行阳离子聚合。 具有腈基、羰基等强吸电子基的单体才能进行阴离子聚合。
离子聚合对单体有较高的选择性 带有1,1-二烷基、烷氧基等推电子基的单体才能进行阳离子聚合。 具有腈基、羰基等强吸电子基的单体才能进行阴离子聚合。 羰基化合物、杂环化合物,大多属离子聚合。 聚合机理和动力学研究不如自由基聚合成熟 聚合条件苛刻,微量杂质有极大影响,聚合重现性差 聚合速率快,需低温聚合,给研究工作造成困难 反应介质的性质对反应也有极大的影响,影响因素复杂 原因

4
离子聚合的发展导致了活性聚合的诞生。这是高分子发展史上的重大转折点。它使高分子合成由必然王国向自由王国迈出了关键的一步。
意义: 1、将难以自由基方式聚合的单体,以离子聚合方式合成新产品; 2、同一单体通过自由基和离子聚合得到的产物的结构与性能不同; 3、在一定程度上可对产物的结构和性能进行设计。 离子聚合的发展导致了活性聚合的诞生。这是高分子发展史上的重大转折点。它使高分子合成由必然王国向自由王国迈出了关键的一步。

5
6.2 阳离子聚合 到目前为止,对阳离子聚合的认识还不很深入。 原因: 阳离子活性很高,极易发生各种副反应,很难获得高分子量的聚合物。
6.2 阳离子聚合 到目前为止,对阳离子聚合的认识还不很深入。 原因: 阳离子活性很高,极易发生各种副反应,很难获得高分子量的聚合物。 引发过程十分复杂,至今未能完全确定。 目前采用阳离子聚合并大规模工业化的产品只有丁基橡胶、聚异丁烯。 阳离子聚合通式可表示如下: 式中A+为阳离子活性中心(碳阳离子,氧鎓离子),难以孤立存在,往往与反离子形成离子对。 B-为反离子,又称抗衡离子 。
,难以孤立存在,往往与反离子形成离子对。 B-为反离子,又称抗衡离子 。")
6
1. 阳离子聚合的烯类单体 从两方面考虑: 具有供电子基的烯类单体原则上可进行阳离子聚合
称为 反离子 从两方面考虑: 供电子基团使双键电子云密度增加,有利于阳离子活性种进攻。 碳阳离子形成后,供电子基团的存在,使碳上电子云稀少的情况有所改变,体系能量有所降低,碳阳离子的稳定性增加。

7
对单体种类进行讨论 (可由热焓-△H判断):
能否聚合成高聚物,还要求: 质子对碳-碳双键有较强的亲合力。 增长反应比其它副反应快,即生成的碳阳离子有适当的稳定性。 对单体种类进行讨论 (可由热焓-△H判断): -烯烃 -△H( kJ/mol) 640 无取代基,不易极化,对质子亲和力小,不能发生阳离子聚合 对质子亲和力较大,有利于反应。 但一个烷基的供电性不强,Rp不快;仲碳阳离子较活泼,容易重排,生成更稳定的叔碳阳离子。
: -烯烃. -△H( kJ/mol) 无取代基,不易极化,对质子亲和力小,不能发生阳离子聚合. 对质子亲和力较大,有利于反应。 但一个烷基的供电性不强,Rp不快;仲碳阳离子较活泼,容易重排,生成更稳定的叔碳阳离子。")
8
故丙烯、丁烯阳离子聚合只能得到低分子油状物。
更高级的α- 烯烃,由于空间位阻效应较大,一般不能通过阳离子聚合得到高分子量聚合物。 两个甲基使双键电子云密度增加很多,易与质子亲合,820 kJ / mol 生成的叔碳阳离子较稳定,可得高分子量的线型聚合物 亚甲基上的氢,受四个甲基的保护,不易夺取,减少了重排、支化等副反应。 是唯一能进行阳离子聚合的-烯烃

9
诱导效应使双键电子云密度降低,氧的电负性较大 共轭效应使双键电子云密度增加,占主导地位。
烷基乙烯基醚 p- 共轭 诱导效应使双键电子云密度降低,氧的电负性较大 共轭效应使双键电子云密度增加,占主导地位。 共振结构使形成的碳阳离子上的正电荷分散而稳定: 能够进行阳离子聚合 但当烷基换成芳基后,由于氧上的未共有电子对也能与芳环形成共轭,分散了双键上的电子云密度,从而使其进行阳离子聚合的活性大大降低。

10
共轭烯烃 如:St,-MeSt,B,IP 电子的活动性强,易诱导极化,既能阳离子聚合,又能阴离子聚合 但聚合活性远不如异丁烯、烷基乙烯醚,工业很少进行这类单体的阳离子聚合生产均聚物。 基本原则: 由于离子聚合的工艺要求较高,故能用自 由基聚合的,尽可能不采用离子聚合。

11
2. 阳离子聚合引发体系及引发作用 阳离子聚合的引发剂都是亲电试剂,即电子接受体 阳离子聚合的引发方式:
1)引发剂生成阳离子,引发单体生成碳阳离子。 2)电荷转移引发:引发剂和单体先形成电荷转 移络合物而后引发。 常用的引发剂:质子酸、 Lewis酸
引发剂生成阳离子,引发单体生成碳阳离子。 2)电荷转移引发:引发剂和单体先形成电荷转. 移络合物而后引发。 常用的引发剂:质子酸、 Lewis酸.")
12
H2SO4,H3PO4,HClO4, CF3COOH,CCl3COOH
质子酸引发 质子酸包括: H2SO4,H3PO4,HClO4, CF3COOH,CCl3COOH 质子酸先电离产生H+,然后与单体加成形成 引发活性中心 活性单体离子对 酸要有足够的强度产生H+,故弱酸不行 酸根的亲核性不能太强,否则会与活性中心结合成共价键而终止,如HCl 条件

13
不同质子酸的酸根的亲核性不同 氢卤酸的X-亲核性太强,不能作为阳离子聚合引发剂,如HCl引发异丁烯 HSO4- H2PO4-的亲核性稍差,可得到低聚体。 HClO4,CF3COOH,CCl3COOH的酸根较弱,可生成高聚物。

14
Lewis酸引发 傅-克(俗称Friedel-Grafts催化剂)反应中的各种金属卤化物,都是电子的接受体,称为Lewis酸。
从工业角度看,是阳离子聚合最重要的引发剂。 Lewis酸包括: 金属卤化物: BF3 , AlCl3, SnCl4 , TiCl4, SbCl5, PCl5, ZnCl2 金属卤氧化物: POCl3,CrO2Cl,SOCl2,VOCl3 绝大部分Lewis酸都需要共(助)引发剂,作为质子或碳阳离子的供给体。
引发剂,作为质子或碳阳离子的供给体。")
15
质子供体:H2O,ROH,HX,RCOOH 碳阳离子供体:RX,RCOX,(RCO)2O
共引发剂有两类: 质子供体:H2O,ROH,HX,RCOOH 碳阳离子供体:RX,RCOX,(RCO)2O 如:无水BF3不能引发无水异丁烯的聚合,加入痕量水,聚合反应立即发生: 引发剂-共引发剂络合物
2O. 如:无水BF3不能引发无水异丁烯的聚合,加入痕量水,聚合反应立即发生: 引发剂-共引发剂络合物.")
16
对于碳阳离子供体供引发剂的情况: RCl 引发剂和共引发剂的组合不同,其活性也不同, 引发剂的活性与接受电子的能力, 即酸性的强弱有关。
BF3 > AlCl3 > TiCl4 > SnCl4 AlCl3 > AlRCl2 >AlR2Cl AlR3 共引发剂的活性视引发剂不同而不同 如异丁烯聚合,BF3为引发剂,共引发剂的活性: 水 :乙酸 :甲醇= 50 :1. 5 :1 SnCl4为引发剂,共引发剂的活性顺序为: HCl>CH3COOH>硝基乙烷>ArOH > H2O>CH3OH>CH3COCH3

17
对于多数聚合,引发剂与共引发剂有一最佳比,在此条件下,Rp最快,分子量最大。
原因: 过量的共引发剂,如水是链转移剂,使链终止,分子量降低。

18
水过量可能生成氧鎓离子,其活性低于引发剂-共引发剂络合物,故Rp下降。
氧鎓离子,活性较低 在工业上,一般采用反应速率较为适中的AlCl3-H2O引发体系。 对有些阳离子聚合倾向很大的单体,可不需要共引发剂,如烷基乙烯基醚。

19
形成的碘阳离子可引发活性较大的单体,如对甲氧基苯乙烯、烷基乙烯基醚等。
其它 包括:I2,高氯酸乙酸酯,电离辐射 如碘分子歧化成离子对,再引发聚合: 形成的碘阳离子可引发活性较大的单体,如对甲氧基苯乙烯、烷基乙烯基醚等。 高氯酸乙酸酯可能是通过酰基正离子与单体加成引发

20
电离幅射引发,可形成单体阳离子自由基,经偶合形成双阳离子活性中心。
幅射引发最大特点:碳阳离子活性中心没有反离子存在

21
电荷转移络合物引发 能进行阳离子聚合的单体都是供电体,当与适当的受电体配合时,能形成电荷转移络合物。在外界能量的作用下,络合物会解离形成阳离子而引发聚合。 如乙烯基咔唑和四腈基乙烯(TCE)的电荷转移引发: [电荷转移络合物]

22
3 阳离子聚合机理 阳离子聚合也是由链引发、链增长和链终止等基元反应组成的。与自由基聚合相比,阳离子聚合有其自身的特点,如快引发、快增长、易转移、难终止,链转移是终止的主要的方式等。 链引发 以引发剂Lewis酸(C)和共引发剂(RH)为例 K
和共引发剂(RH)为例. K.")
23
阳离子引发活化能为Ei = 8.4~21 kJ/mol(自由基聚合的Ei = 105~150 kJ/mol),引发极快,瞬间完成。
若第二步是速率控制反应 特点: 阳离子引发活化能为Ei = 8.4~21 kJ/mol(自由基聚合的Ei = 105~150 kJ/mol),引发极快,瞬间完成。
,引发极快,瞬间完成。")
24
链增长 引发反应生成的碳阳离子活性中心与反离子始终构成离子对,单体分子不断插入其中而增长。 特点: 增长速率为
增长速率快,活化能(Ep=8.4~21kJ/mol)低,几乎与引发同时完成。
低,几乎与引发同时完成。")
25
增长活性中心为一离子对,单体按头尾结构插入离子对而增长,对链节构型有一定的控制能力。离子对的紧密程度与溶剂、反离子性质、温度等有关,对聚合速率、分子量和构型有较大影响。
伴有分子内重排反应,异构成更稳定的结构 如 3-甲基-1-丁烯聚合产物有两种结构: 重排通常是通过电子或个别原子的转移进行的。 这种通过增长链碳阳离子发生重排的聚合反应称为异构化聚合或氢转移聚合。

26
链转移和链终止 离子聚合的增长活性中心带有相同的电荷,不能双分子终止,只能发生链转移终止或单基终止,也可人为添加终止剂终止。无凝胶效应。 这一点与自由基聚合显著不同。 自由基聚合的链转移一般不终止动力学链,而阳离子聚合的链转移则有可能终止动力学链。因此阳离子聚合的链终止可分为动力学链不终止的链终止反应和动力学链终止的链终止反应两类。

27
动力学链不终止 向单体转移终止 活性链向单体转移,生成的大分子含有不饱和端基,同时再生出活性单体离子对,动力学链不终止。

28
向单体转移常数CM,约为10-1~10-2,比自由基聚合(10-4~10-5)大,易发生转移反应。
反应通式为 转移速率为: 特点: 向单体转移是主要的链终止方式之一。 向单体转移常数CM,约为10-1~10-2,比自由基聚合(10-4~10-5)大,易发生转移反应。 是控制分子量的主要因素,也是阳离子聚合必须低温反应的原因。 例如,异丁烯的聚合,T = 0~-40℃,Mn <5万,T = -100 ℃, Mn = 5 万~500万
大,易发生转移反应。 是控制分子量的主要因素,也是阳离子聚合必须低温反应的原因。 例如,异丁烯的聚合,T = 0~-40℃,Mn <5万,T = -100 ℃, Mn = 5 万~500万.")
29
向反离子转移终止或自发终止 增长链重排导致活性链终止,再生出引发剂-共引发剂络合物,继续引发单体,动力学链不终止。 反应通式: 自发终止速率:

30
反离子亲核性足够强时会与增长的碳阳离子以共价键结合而终止。
动力学链终止 与反离子加成终止 反离子亲核性足够强时会与增长的碳阳离子以共价键结合而终止。 例如三氟乙酸引发的苯乙烯聚合:

31
阳离子聚合自身不容易终止,通过添加水、醇、酸、醚、胺、醌等终止剂可使聚合终止。
与反离子中的阴离子部分加成终止 使引发剂-共引发剂比例改变。 添加终止剂 阳离子聚合自身不容易终止,通过添加水、醇、酸、醚、胺、醌等终止剂可使聚合终止。

32
链终止剂 XA 主要有: 水、醇、酸、酐、酯、醚、胺、醌 形成的氧鎓离子活性低,不能再引发聚合。

33
苯醌既是自由基聚合的阻聚剂,又对阳离子聚合起阻聚作用。
阳离子聚合机理的特点: 快引发,快增长,易转移,难终止

34
4 阳离子聚合动力学 聚合体系多为非均相 共引发剂、微量杂质对聚合速率影响很大 聚合速率快,数据重现性差 真正的终止反应不存在,稳态假定难以建立 比自由基聚合研究困难 离子聚合无双基终止,无自动加速现象,往往以低活性的 SnCl4 为引发剂,向反离子转移作为终止方式时的聚合作为典型进行讨论。各基元反应的动力学方程为:

35
聚合速率 引发: 引发剂引发生成碳阳离子的反应是控制速率反应 增长 终止

36
建立稳态 Rp 对引发剂、共引发剂浓度呈一级反应, 对单体浓度呈二级反应 自发终止时,引发剂浓度为常数,
而向反离子加成时,引发剂浓度下降。

37
讨论: 是假定引发过程中引发剂引发单体生成碳阳离子的反应是控制速率反应,因此Ri与单体浓度有关。
若引发剂与共引发剂的反应是慢反应,则 Ri与单体浓度无关,Rp与单体浓度一次方成正比。 该动力学方程也适合于与反离子加成终止、向单体转移终止(表达式有变动), 但不宜推广到其它聚合体系。 离子聚合无双基终止,不会出现自动加速现象
, 但不宜推广到其它聚合体系。 离子聚合无双基终止,不会出现自动加速现象.")
38
在阳离子聚合中,向单体转移和向溶剂转移是主要的终止方式,虽然转移后聚合速率不变,但聚合度降低。转移速率方程为:
参照自由基聚合,可将阳离子聚合物的聚合度表达为: 单基终止

39
单基终止为主要终止方式时 向单体链转移为主要终止方式时 向溶剂转移终止时

40
例如,聚异丁烯的制备采用在CH3Cl溶剂中的阳离子聚合。终止方式有向单体链转移和向溶剂链转移两种,取决于温度的影响。聚合温度低于-100℃,主要向单体转移终止;聚合温度高于100℃,主要向溶剂转移终止。 AlCl3引发异丁烯聚合时温度与聚合度的关系

41
Rp阳 >> Rp自 各基元反应速率常数 速率常数 阳离子聚合 自由基聚合 kp ( L / mol•s) 7. 6 100
速率常数 阳离子聚合 自由基聚合 kp ( L / mol•s) kt ×10-2 (s-1) ( l / mol•s) kp / kt kp / kt1/2 10-2 活性种浓度 [C*] ~ 10- [ M •] ~ 10-8 Rp阳 >> Rp自
 kt 4.9×10-2 (s-1) 10 7 ( l / mol•s) kp / kt 102 kp / kt1/2 10-2. 活性种浓度 [C*] ~ 10-3 [ M •] ~ 10-8. Rp阳 >> Rp自.")
42
5 影响阳离子聚合的因素 反应介质(溶剂)的影响 活性中心离子对的形态 在不同溶剂中, 活性中心离子和反离子有不同形态
共价键 紧密离子对 溶剂分隔的离子对 自由离子 平衡离子对 大多数聚合活性种处于平衡离子对和自由离子状态 kp(+) :自由离子增长速率常数 kp() :离子对增长速率常数 kp(+) > kp() 1~3个数量级 阳离子聚合以离子对为主,但只占一小部分的自由离子对总速率的贡献很大。
 :自由离子增长速率常数. kp() :离子对增长速率常数. kp(+) > kp() 1~3个数量级. 阳离子聚合以离子对为主,但只占一小部分的自由离子对总速率的贡献很大。")
43
溶剂的极性和溶剂化能力大,自由离子和疏松离子对的比例增加,聚合速率和分子量增大。
一般情况下,离子对为松对时的聚合速率和聚合度均较大。 溶剂的极性和溶剂化能力的影响 溶剂的极性和溶剂化能力大,自由离子和疏松离子对的比例增加,聚合速率和分子量增大。 溶剂的极性常用介电常数 表示。 ,表观kp 但要求:不能与中心离子反应;在低温下溶解反应物保持流动性。故采用低极性溶剂,如卤代烷:四氯化碳,氯仿,二氯乙烷;硝基化合物如硝基甲烷和硝基苯。芳烃类化合物如苯及甲苯等较少使用。

44
反离子的影响 反离子的亲核性 亲核性强,易与碳阳离子结合,使链终止。如Cl- 一般不宜作为反离子。 反离子的体积
反离子的体积 体积大,离子对疏松,聚合速率大。 例如,用I2、SnCl4-H2O、HClO4引发苯乙烯在1,2 -二氯乙烷中25℃下的阳离子聚合,聚合速率常数分别为0.003、0.42、1.70 L/mol.s。

45
温度的影响 对聚合速率的影响 综合活化能为正值时,温度降低,聚合速率减小 综合活化能为负值时,温度降低,聚合速率加快
综合速率常数 综合活化能为正值时,温度降低,聚合速率减小 综合活化能为负值时,温度降低,聚合速率加快 但综合活化能的绝对值较小,温度影响也较小。

46
对聚合度的影响 Et 或 Etr,M 一般总大于Ep,综合活化能为负值,为-12.5 ~ -29 kJ / mol。 因此,聚合度随温度降低而增大,这是阳离子聚合在较低温度下进行聚合的原因。同时温度低还可以减弱副反应。

47
温度影响离子对与自由离子的比例和聚合速度,温度升高,离子对比例增加,速度变慢,分子量变小。
离子对的解离是放热反应,降低温度有利于生成自由离子。 在阳离子聚合反应中,链终止的主要方式为向单体转移,CM大,温度升高, CM增加,聚合物的分子量变小。所以阳离子聚合控制温度最为重要。

48
6 阳离子聚合应用实例 丁基橡胶 异丁烯和少量异戊二烯(1~6%)为单体, AlCl3为引发剂,氯甲烷为稀释剂, 在-100℃下聚合, 瞬间完成,分子量达20万以上。 丁基橡胶冷却时不结晶,-50℃柔软,耐候,耐臭氧,气密性好,主要用作内胎。
为单体, AlCl3为引发剂,氯甲烷为稀释剂, 在-100℃下聚合, 瞬间完成,分子量达20万以上。 丁基橡胶冷却时不结晶,-50℃柔软,耐候,耐臭氧,气密性好,主要用作内胎。 .")
49
聚异丁烯 AlCl3为引发剂, 氯甲烷为溶剂,在0 ~-40℃聚合, 得低分子量(<5万)聚异丁烯,主要用于粘结剂、密封材料等;在-100℃下聚合,得高分子量产物(5万~100万),主要用作橡胶制品。 主要通过改变温度调节分子量。

50
聚乙烯基醚: 聚乙烯基甲醚为水溶性高分子,水溶液在纺织工业中用作打浆剂。

51
6.3 阴离子聚合 在链式聚合反应中,活性中心为阴离子的聚合反应。常用的引发剂有碱金属、丁基锂等亲核试剂。 阴离子聚合反应的通式可表示如下:
6.3 阴离子聚合 在链式聚合反应中,活性中心为阴离子的聚合反应。常用的引发剂有碱金属、丁基锂等亲核试剂。 阴离子聚合反应的通式可表示如下: 其中B-为阴离子活性中心,A+为反离子,一般为金属离子。与阳离子聚合不同,阴离子聚合中,活性中心可以是自由离子、离子对,以及处于缔合状态的阴离子。

52
1.阴离子聚合的烯类单体 具有吸电子取代基的烯类单体原则上可以进行阴离子聚合。 能否聚合取决于两种因素: 是否具有-共轭体系
吸电子基团并具有-共轭体系,能够进行阴离子聚合,如AN、MMA、硝基乙烯。 吸电子基团并不具有-共轭体系,则不能进行阴离子聚合,如VC、VAc( p- )。 与吸电子能力有关 +e 值越大,吸电子能力越强,易进行阴离子聚合。
。 与吸电子能力有关. +e 值越大,吸电子能力越强,易进行阴离子聚合。")
53
2. 阴离子聚合的引发剂和引发反应 阴离子聚合的引发剂是电子给体(亲核试剂),属碱类物质(碱金属,有机金属化合物以及三级胺等)。
根据引发机理可分为: 电子转移引发和阴离子引发两类。

54
碱金属—电子转移引发 Li、Na、K等碱金属原子最外层仅一个价电子,容易转移给单体或其他化合物,生成单体自由基-阴离子,并进而形成双阴离子引发聚合。 电子直接转移引发 单体自由基-阴离子 双阴离子活性中心 碱金属不溶于溶剂,属非均相体系;引发反应较慢,引发剂利用率低; 产物分子量分布宽。

55
碱金属将电子转移给中间体,形成自由基-阴离子,再将活性转移给单体,如萘钠在THF中引发St
电子间接转移引发 碱金属将电子转移给中间体,形成自由基-阴离子,再将活性转移给单体,如萘钠在THF中引发St THF (红色) (绿色) 双阴离子 萘钠在极性溶剂中是均相体系,碱金属的利用率高。
 (绿色) 双阴离子. 萘钠在极性溶剂中是均相体系,碱金属的利用率高。")
56
有机金属化合物-阴离子引发 金属氨基化合物 金属胺基化合物、金属烷基化合物和格利雅试剂等。 是研究得最早的一类引发剂。
主要有 NaNH2-液氨、KNH2 -液氨体系可以自由 阴离子形式引发聚合。 形成自由阴离子 单阴离子 这类引发剂的活性太大,聚合不易控制,故目前已不使用。

57
金属烷基化合物 金属的电负性 引发活性与金属的电负性有关。金属与碳的电负性相差越大,越容易形成离子。 如丁基锂以离子对方式引发
K Na Li Mg Al 电负性 ~ 金属-碳键 K-C Na-C Li-C Mg-C Al-C 键的极性 有离子性 极性共价键 极性弱 极性更弱 引发作用 活泼引发剂 常用引发剂 不能直接引发 不能 金属的电负性 如丁基锂以离子对方式引发 制成格氏试剂,引发活泼单体

58
丁基锂

59
其它亲核试剂 中性亲核试剂,如R3P、R3N、ROH、H2O等,都有未共用的电子对,在引发和增长过程中生成电荷分离的两性离子。
只能引发非常活泼的单体 3 阴离子聚合引发剂与单体的匹配 阴离子聚合的单体和引发剂的活性各不相同,并具有选择性。只有某些引发剂才能引发某些单体。

60
基本原则为:活性大的引发剂可引发活性活从小至大的种单体;而引发活性小的引发剂,只能引发活性大的单体,见图5-2。
图5-2 阴离子聚合引发剂和单体的匹配

61
4. 活性阴离子聚合 机理 阴离子聚合的特点: 快引发,慢增长,无终止和无转移。 (1)引发反应
阴离子聚合的引发活性种可以是离子对和自由离子。这与溶剂的极性有关。 极性溶剂:自由离子;非极性溶剂:离子对, 也有同时存在的情况。 (2)增长反应 在增长反应中,单体的加成方向受离子对的限制,产物的立构规整性较自由基聚合强,但不能控制。
增长反应. 在增长反应中,单体的加成方向受离子对的限制,产物的立构规整性较自由基聚合强,但不能控制。")
62
(3)终止反应 离子聚合无双基终止。阳离子聚合主要通过链转移终止。而阴离子聚合连链转移反应都很难发生,因此实际上不存在终止反应。增长反应中的活性链直到单体完全耗尽仍可保持活性,因此有“活性聚合”的概念。 每一活性中心所连接的单体数基本相等,分子量等于单体摩尔数除以引发剂摩尔数,且比较均匀,分布窄。 1956年,Swarc 采用萘-钠引发体系,以THF为溶剂进行苯乙烯阴离子聚合,首次发现活性聚合物。

63
活性聚合物 阴离子聚合在适当条件下(体系非常纯净;单体为非极性共轭双烯),可以不发生链终止或链转移反应,活性链直到单体完全耗尽仍可保持聚合活性。 这种单体完全耗尽仍可保持聚合活性的聚合物链阴离子称为“ 活性高分子”(Living Polymer)。 实验证据 萘钠在THF中引发苯乙烯聚合,碳阴离子增长链为红色,直到单体100%转化,红色仍不消失。 重新加入单体,仍可继续链增长(放热),红色消退非常缓慢,几天~几周。
,红色消退非常缓慢,几天~几周。")
64
阴离子聚合不存在真正的链终止反应。 形成活性聚合物的原因 离子聚合无双基终止 反离子为金属离子,不能加成终止
从活性链上脱除氢负离子H-进行链转移困难,所需能量较高(主要原因) 最终仍可脱H-终止,可能发生下述反应: 阴离子聚合不存在真正的链终止反应。 氢化纳活性较大,可再度引发聚合
 最终仍可脱H-终止,可能发生下述反应: 阴离子聚合不存在真正的链终止反应。 氢化纳活性较大,可再度引发聚合.")
65
烯丙基氢 1, 3-二苯基烯丙基阴离子 由于共轭效应,很稳定,无反应活性

66
在聚合末期,加入链转移剂(水、醇、酸、胺)可使活性聚合物终止。 有目的的加入CO2、环氧乙烷、二异氰酸酯可获得指定端基聚合物。
端羧基化反应 端羟基化反应

67
端胺基化反应

68
聚合速率 Rp = kp [M–] [M] 式中 kp 表观速率常数 [M-] 阴离子活性增长中心的总浓度 该式成立条件:
由于阴离子聚合为活性聚合,聚合前引发剂快速全部转变为活性中心,且活性相同。增长过程中无再引发反应,活性中心数保持不变,无终止,因此可写出速率方程: Rp = kp [M–] [M] 式中 kp 表观速率常数 [M-] 阴离子活性增长中心的总浓度 该式成立条件: 无杂质的活性聚合,且引发快于增长反应 即在开始聚合前,引发剂已定量地离解成活性中心,则阴离子活性中心的浓度等于引发剂的浓度,[M-] = [ C] 如果Ri≤Rp,[M-] 将不断降低,则上式不能采用。
![聚合速率 Rp = kp [M–] [M] 式中 kp 表观速率常数 [M-] 阴离子活性增长中心的总浓度 该式成立条件:](http://slidesplayer.com/slide/11155198/59/images/68/%E8%81%9A%E5%90%88%E9%80%9F%E7%8E%87+Rp+%3D+kp+%5BM%E2%80%93%5D+%5BM%5D+%E5%BC%8F%E4%B8%AD+kp+%EF%82%BE%E8%A1%A8%E8%A7%82%E9%80%9F%E7%8E%87%E5%B8%B8%E6%95%B0+%5BM%EF%BC%8D%5D+%EF%82%BE%E9%98%B4%E7%A6%BB%E5%AD%90%E6%B4%BB%E6%80%A7%E5%A2%9E%E9%95%BF%E4%B8%AD%E5%BF%83%E7%9A%84%E6%80%BB%E6%B5%93%E5%BA%A6+%E8%AF%A5%E5%BC%8F%E6%88%90%E7%AB%8B%E6%9D%A1%E4%BB%B6%EF%BC%9A.jpg "由于阴离子聚合为活性聚合,聚合前引发剂快速全部转变为活性中心,且活性相同。增长过程中无再引发反应,活性中心数保持不变,无终止,因此可写出速率方程: Rp = kp [M–] [M] 式中 kp 表观速率常数. [M-] 阴离子活性增长中心的总浓度. 该式成立条件: 无杂质的活性聚合,且引发快于增长反应. 即在开始聚合前,引发剂已定量地离解成活性中心,则阴离子活性中心的浓度等于引发剂的浓度,[M-] = [ C] 如果Ri≤Rp,[M-] 将不断降低,则上式不能采用。")
69
转化率达100%时,单体全部平均分配到每个活性端基上。
阴离子的聚合速率比自由基聚合大104~107倍 从kp值比较,两者相近。 从活性中心浓度比较: [M-] -3 ~ 10-2 mol / L [M•] -9 ~ 10-7 mol / L [M-] > [M•] ~ 倍 聚合度 在下列条件下: 引发剂全部很快地转变成活性中心 搅拌良好,单体分布均匀,所有链增长同时开始 无链转移和链终止反应 解聚可忽略 转化率达100%时,单体全部平均分配到每个活性端基上。

70
活性聚合物的平均聚合度等于单体浓度与大分子活性链数之比:
=活性端基浓度/n 式中 [C] 引发剂浓度 n 每个引发剂分子上的活性中心数 双阴离子 n = 2 单阴离子 n = 1 通过定量计算加入引发剂和单体,从而得到预期聚合度和窄分子量分布的聚合反应称为化学计量聚合。

71
活性阴离子聚合的分子量分布服从Flory分布或Poissen分布,x-聚体的摩尔分率为:
得到的产物的分子量分布很窄,接近单分散。 如St在THF中聚合,分子量分布指数= ~ 1. 12 可作标准样品。但仍存在一定分散性,原因: 反应过程中很难使引发剂分子与单体完全混合均匀,即每个活性中心与单体混合的机会总是有些差别; 不可能将体系中的杂质完全清除干净。

72
增长速率常数及其影响因素 (1) 溶剂和反离子性质的影响
(1) 溶剂和反离子性质的影响 如果溶剂和反离子性质不同,则离子对的松紧程度可以差别很大,影响到单体插入增长的速率。增长活性种可以处于各种状态,如共价键、紧离子对、松离子对、自由离子等,彼此互相平衡。聚合速率是处于平衡状态的离子对和自由离子共同作用的结果。
 溶剂和反离子性质的影响. 如果溶剂和反离子性质不同,则离子对的松紧程度可以差别很大,影响到单体插入增长的速率。增长活性种可以处于各种状态,如共价键、紧离子对、松离子对、自由离子等,彼此互相平衡。聚合速率是处于平衡状态的离子对和自由离子共同作用的结果。")
73
介电常数,表示溶剂极性的大小,溶剂极性越大,活性链离子与反离子的离解程度越大,自由离子越多。
溶剂的性质可用两个物理量表示: 介电常数,表示溶剂极性的大小,溶剂极性越大,活性链离子与反离子的离解程度越大,自由离子越多。 电子给予指数,反映了溶剂的给电子能力。 溶剂的给电子能力强,对阳离子的溶剂化作用越强,离子对也越分开。 溶剂能导致活性中心的形态结构及活性发生变化。

74
总聚合速率是离子对 P-C+ 和自由离子 P- 聚合速率之和:
其中活性种总浓度: 两活性种平衡常数:

75
一般情况下: 可推出 在多数情况下,离子对解离程度很低,

76
通常 比 大102~103倍。在溶剂化能力较大的溶剂中(如四氢呋喃),反离子体积越大,解离程度越低,越易形成紧对,由锂到铯,
以kp对[M-]-1/2作图,可得直线,截距为 ,斜率为 。再通过电导法测得平衡常数 K 后,就可以求得 。 反离子与溶剂化程度有关 通常 比 大102~103倍。在溶剂化能力较大的溶剂中(如四氢呋喃),反离子体积越大,解离程度越低,越易形成紧对,由锂到铯, 随反离子半径增加而减小。 而在溶剂化能力小的二氧六环溶剂中,离子对不易电离,也不易使反离子溶剂化,因此离子对增长速率常数很小,同时随反离子半径大,离子对间距增大,单体易插入,结果 随反离子半径增加而增大。
,反离子体积越大,解离程度越低,越易形成紧对,由锂到铯, 随反离子半径增加而减小。 而在溶剂化能力小的二氧六环溶剂中,离子对不易电离,也不易使反离子溶剂化,因此离子对增长速率常数很小,同时随反离子半径大,离子对间距增大,单体易插入,结果 随反离子半径增加而增大。")
77
(2)温度对增长速率常数的影响 活性聚合的活化能一般为较小的正值(8~20 kJ/mol),因此聚合速率随温度升高略有增加,但不敏感。 升高温度可使离子对和自由离子的聚合速率常数提高,但使两者的平衡常数降低。 不同性质的溶剂,温度对聚合速率常数的影响不同。

78
溶剂化能力较弱的溶剂(如二氧六环)中,离子对解离能力较弱,温度对K的影响较小,增长速率主要取决于离子对,表观活化能较大,温度对聚合速率影响较大。
溶剂化能力较强的溶剂(如四氢呋喃)中,离子对解离能力较大,温度对K的影响也较大。因此温度对K和 、 的影响抵消,表观活化能较低,则温度对聚合速率影响较小。
中,离子对解离能力较大,温度对K的影响也较大。因此温度对K和 、 的影响抵消,表观活化能较低,则温度对聚合速率影响较小。")
79
活性聚合物的应用 (1)测定阴离子聚合增长速率常数 (2)合成均一分子量的聚合物 目前合成均一特定分子量的唯一方法,为GPC提供标准样品。
单分散聚苯乙烯的制备: 丁基锂/正庚烷/St; -70℃; 分子量分布1.01。 GPC 测试参比样品。

80
指分子链两端都带有活性官能团的聚合物,两个官能团遥遥位于分子链的两端,就象两个爪子,故称为遥爪聚合物。
(3)制备带有特殊官能团的遥爪聚合物 遥爪聚合物: 指分子链两端都带有活性官能团的聚合物,两个官能团遥遥位于分子链的两端,就象两个爪子,故称为遥爪聚合物。 前述制备端基官能团的方法,如果是双阴离子聚合,则可得到遥爪聚合物。
制备带有特殊官能团的遥爪聚合物. 遥爪聚合物: 指分子链两端都带有活性官能团的聚合物,两个官能团遥遥位于分子链的两端,就象两个爪子,故称为遥爪聚合物。 前述制备端基官能团的方法,如果是双阴离子聚合,则可得到遥爪聚合物。")
81
利用活性聚合,先制得一种单体的活性链,然后加入另一种单体,可得到所希望链段长度的嵌段共聚物。
(4)制备嵌段共聚物 利用活性聚合,先制得一种单体的活性链,然后加入另一种单体,可得到所希望链段长度的嵌段共聚物。 工业上已经用这种方法合成了St-B、St-B-St两嵌段和三嵌段共聚物。 这种聚合物在室温具有橡胶的弹性,在高温又具有塑料的热塑性,可用热塑性塑料的加工方法加工,故称为热塑弹性体。 并非所有的活性链都可引发另一单体,能否进行上述反应,取决于M1-和M2的相对碱性,即M1-给电子能力和M2的亲电子能力。
制备嵌段共聚物. 利用活性聚合,先制得一种单体的活性链,然后加入另一种单体,可得到所希望链段长度的嵌段共聚物。 工业上已经用这种方法合成了St-B、St-B-St两嵌段和三嵌段共聚物。 这种聚合物在室温具有橡胶的弹性,在高温又具有塑料的热塑性,可用热塑性塑料的加工方法加工,故称为热塑弹性体。 并非所有的活性链都可引发另一单体,能否进行上述反应,取决于M1-和M2的相对碱性,即M1-给电子能力和M2的亲电子能力。")
82
实验发现: 对于单体,存在下列共轭酸碱平衡:
Kd是电离平衡常数,用 pKd = -log Kd表示单体相对碱性的大小, pKd值越大,单体的碱性越大。 实验发现: pKd值大的单体形成链阴离子后,能引发pKd值小的单体,反之不能。 如 pKd值:St 40 ~ 42 ; MMA 24

83
(5)制备星型聚合物 pKd值同一级别的单体也有方向性 St-能引发B,反之相对困难,因为B-比St-更稳定
不能 pKd值同一级别的单体也有方向性 St-能引发B,反之相对困难,因为B-比St-更稳定 (5)制备星型聚合物 通过偶联剂将聚合物链阴离子连接起来,可获得星型聚合物。
制备星型聚合物. 通过偶联剂将聚合物链阴离子连接起来,可获得星型聚合物。")
84
5 丁基锂的缔合现象和定向聚合作用 缔合现象 烷基锂在非极性溶剂中存在缔合现象,单分子丁基锂与缔合分子处于平衡中,只有单分子才能引发聚合。
5 丁基锂的缔合现象和定向聚合作用 丁基锂是目前应用最广的阴离子聚合引发剂。实践中发现若溶剂体系选择不当,丁基锂的引发活性很低,这可能是由于丁基锂的缔合作用引起。 丁基锂在特定条件下对聚合产物具有定向作用。 缔合现象 烷基锂在非极性溶剂中存在缔合现象,单分子丁基锂与缔合分子处于平衡中,只有单分子才能引发聚合。 若浓度很低<10-4M,基本不缔合; 浓度高时,在苯中,正丁基锂为6 分子缔合体,活性链为2 分子缔合体。 (C4H9Li) C4H9 Li; 若加入四氢呋喃,缔合程度小,聚合反应加快。 K1 K2
6 6C4H9 Li; 若加入四氢呋喃,缔合程度小,聚合反应加快。 K1. K2.")
85
定向聚合作用 常温下,丁二烯和异戊二烯自由基聚合,10%-20%,为1,4-顺式。 非极性溶剂中: 烷基锂引发阴离子聚合:丁二烯,35% 1,4-顺式; 异戊二烯,94%1,4-顺式(合成天然橡胶) 极性溶剂中: 1,4-顺式结构显著降低。 丁二烯,80% 1,2结构; 异戊二烯,75% 3,4-顺式。

86
6 阴离子聚合的立体化学 在阴离子聚合中,由于链增长活性中心与抗衡阳离子之间存在相互作用,单体与链增长活性中心加成时,其取向会受到这种相互作用的影响,因而具有一定的立体定向性。其定向程度取决于抗衡阳离子与链增长活性中心的离解程度。

87
极性溶剂中:链增长活性中心与抗衡阳离子表现为溶剂分离离子对或自由离子,两者之间的相互作用较弱,单体与链增长活性中心加成时,主要受立体因素影响而采取立体阻碍最小的方式加成,有利于得到间同立构产物:

88
非极性溶剂中:链增长活性中心与抗衡阳离子表现为紧密离子对,相互间作用较强,单体与链增长活性中心加成是主要受这种相互作用的影响,有利于获得全同立构高分子。
随着溶剂极性的提高或金属被其它弱配位能力的金属替代,产物的立体规整性下降。

89
温度 对离子对和自由离子平衡的影响 K 由于离子对的离解是放热反应,随着温度的降低,离解平衡常数(K值)变大,有利于平衡向右进行,即增加自由离子浓度,从而影响产物的立体规整性。
变大,有利于平衡向右进行,即增加自由离子浓度,从而影响产物的立体规整性。")
90
6.4 离子聚合与自由基聚合的比较 引发剂种类 自由基聚合: 采用受热易产生自由基的物质作为引发剂
6.4 离子聚合与自由基聚合的比较 引发剂种类 自由基聚合: 采用受热易产生自由基的物质作为引发剂 偶氮类 过氧类 氧化还原体系 引发剂的性质只影响引发反应,用量影响 Rp和 离子聚合 采用容易产生活性离子的物质作为引发剂 阳离子聚合:亲电试剂,主要是Lewis酸,需共引发剂 阴离子聚合:亲核试剂,主要是碱金属及金属有机化合物

91
单体结构 自由基聚合 离子聚合:对单体有较高的选择性 引发剂中的一部分,在活性中心近旁成为反离子
其形态影响聚合速率、分子量、产物的立构规整性。 单体结构 带有弱吸电子基的乙烯基单体 共轭烯烃 自由基聚合 离子聚合:对单体有较高的选择性 阳离子聚合:带有强推电子取代基的烯类单体 共轭烯烃(活性较小) 阴离子聚合:带有强吸电子取代基的烯类单体 共轭烯烃 环状化合物、羰基化合物
 阴离子聚合:带有强吸电子取代基的烯类单体. 共轭烯烃. 环状化合物、羰基化合物.")
92
溶剂的影响 自由基聚合 离子聚合 向溶剂链转移,降低分子量 笼蔽效应,降低引发剂效率 f 溶剂加入,降低了[M],Rp略有降低
水也可作溶剂,进行悬浮、乳液聚合 溶剂的极性和溶剂化能力,对活性种的形态有较大影响:离子对、自由离子 影响到 Rp、Xn 和产物的立构规整性 溶剂种类 离子聚合 阳:卤代烃、CS2、液态SO2、CO2 阴:液氨、醚类 (THF、二氧六环)
![溶剂的影响 自由基聚合 离子聚合 向溶剂链转移,降低分子量 笼蔽效应,降低引发剂效率 f 溶剂加入,降低了[M],Rp略有降低](http://slidesplayer.com/slide/11155198/59/images/92/%E6%BA%B6%E5%89%82%E7%9A%84%E5%BD%B1%E5%93%8D+%E8%87%AA%E7%94%B1%E5%9F%BA%E8%81%9A%E5%90%88+%E7%A6%BB%E5%AD%90%E8%81%9A%E5%90%88+%E5%90%91%E6%BA%B6%E5%89%82%E9%93%BE%E8%BD%AC%E7%A7%BB%EF%BC%8C%E9%99%8D%E4%BD%8E%E5%88%86%E5%AD%90%E9%87%8F+%E7%AC%BC%E8%94%BD%E6%95%88%E5%BA%94%EF%BC%8C%E9%99%8D%E4%BD%8E%E5%BC%95%E5%8F%91%E5%89%82%E6%95%88%E7%8E%87+f+%E6%BA%B6%E5%89%82%E5%8A%A0%E5%85%A5%EF%BC%8C%E9%99%8D%E4%BD%8E%E4%BA%86%5BM%5D%EF%BC%8CRp%E7%95%A5%E6%9C%89%E9%99%8D%E4%BD%8E.jpg "水也可作溶剂,进行悬浮、乳液聚合. 溶剂的极性和溶剂化能力,对活性种的形态有较大影响:离子对、自由离子. 影响到 Rp、Xn 和产物的立构规整性. 溶剂种类. 离子聚合. 阳:卤代烃、CS2、液态SO2、CO2. 阴:液氨、醚类 (THF、二氧六环)")
93
反应温度 聚合机理 自由基聚合:取决于引发剂的分解温度,50 ~ 80 ℃。 离子聚合:引发活化能很小。
为防止链转移、重排等副反应,在低温聚合, 阳离子聚合常在-70 ~100 ℃进行。 聚合机理 双基偶合 双基歧化 Rp [I] 1/2 自由基聚合:多为双基终止 无自加速现象 Rp [C] 离子聚合:具有相同电荷,不能双基终止 阳:向单体、反离子、链转移剂终止 阴:往往无终止,活性聚合物,添加其它试剂终止

94
阻聚剂种类 机理特征: 自由基聚合:慢引发、快增长、速终止、可转移 阳离子聚合:快引发、快增长、易转移、难终止
阴离子聚合:快引发、慢增长、无终止 阻聚剂种类 自由基聚合:氧、DPPH、苯醌 阳离子聚合:极性物质水、醇,碱性物质,苯醌 阴离子聚合:极性物质水、醇,酸性物质,CO2 问题:有DPPH和苯醌两种试剂,如何区别三种反应?

95
6.5 离子共聚 自由基共聚物组成方程,也适用于离子共聚,但两类共聚也有差异。 1 离子共聚对单体有较高选择性,能进行离子共聚的单体对比自由基共聚少得多。 同一对单体用不同机理的引发体系进行共聚时,竞聚率和共聚物组成会有很大差别。 2 离子共聚的单体极性一般相近,有理想共聚的倾向,r1r2约等于1,少数r1r2 >1。 有交替倾向的单体难进行离子共聚,因为这类单体对极性差别很大。 3 溶剂和反离子的性质和温度对单体活性和竞聚率有很 大影响,从而影响共聚物的组成。

96
取代基对单体活性的影响 电子效应 空间位阻效应 带供电子基团的烯类单体有利于阳离子聚合。活性取决于取代基供电子或吸电子能力大小。
阳离子聚合单体活性次序一般情况: 乙烯基醚类 > 异丁烯> 苯乙烯> 异戊二烯 带吸电子基团的烯类单体有利于阴离子聚合。 吸电子取代基使单体活性增加顺序: -CN>-COOR>-C6H5>-CH=CH2>-H 空间位阻效应 取代基的空间位置不同,对单体离子聚合的活性有显著影响。

97
溶剂和反离子的影响 溶剂极性和溶剂化能力、反离子性质对单体离子共聚的竞聚率有很大影响。 温度的影响 温度对离子共聚竞聚率的影响很大,对不同单体的影响也不一。 自由基共聚中,不同单体对同一自由基增长活化能的差值很小,温度对竞聚率的影响很小。升高温度,使两竞聚率向1逼近。 离子共聚中,温度对竞聚率影响较大,且影响程度不一致。

98
6.6 开环聚合 开环聚合:环状单体在某种引发剂作用下形成线性聚合物的过程。 开环聚合—无小分子副产物产生(缩聚);无双键的断裂(加聚);环张力的释放是开环聚合的推动力。 在聚合反应旧的分类中,与加聚、缩聚并列,为第三大类聚合反应。 按机理考虑:大部分开环聚合属于连锁机理的离子聚合。小部分属于逐步聚合。 单体:环醚、环缩醛、环酯、环酰胺、环硅烷等。环氧乙烷、环氧丙烷、己内酰胺、三聚甲醛等的开环聚合都是重要的工业化开环聚合反应。

99
1 环烷烃开环聚合热力学 开环的影响因素:环的大小,构成环的元素(碳环或杂环)、环上取代基。
热力学:键的变形程度愈大,环的张力能和聚合热也愈大,聚合自由焓负值更小,则环的稳定性愈低,愈易开环聚合。 此外,双官能度单体线性缩聚还有环化倾向。这些现象都取决于环和线性结构的相对稳定性,属于热力学因素。 环的大小对环稳定性和开环倾向的影响,在热力学上可由键角大小、键的变形程度、环的张力能、聚合热、聚合自由焓等作定性或半定量的判断。 不同大小环的热力学稳定次序: 3,4<<5,7~11<12以上,6

100
按碳的四面体结构,C—C—C键角为109°28’,而环状化合物的键角有不同程度的变形,因此产生张力。
三、四元环烷烃由键角变化引起的环张力很大(三元环60°,四元环90 °),环不稳定而易开环聚合;五元环键角接近正常键角(108 °) ,张力较小,环较稳定。五元以上环可以不处于同一平面使键角变形趋于零而难开环。六元环烷烃通常呈椅式结构,键角变形为0,不能开环聚合。八元以上的环有跨环张力,即环上的氢或其他取代基处于拥挤状态所造成的斥力,聚合能力较强。十一元以上环的跨环张力消失,环较稳定,不易聚合。
,环不稳定而易开环聚合;五元环键角接近正常键角(108 °) ,张力较小,环较稳定。五元以上环可以不处于同一平面使键角变形趋于零而难开环。六元环烷烃通常呈椅式结构,键角变形为0,不能开环聚合。八元以上的环有跨环张力,即环上的氢或其他取代基处于拥挤状态所造成的斥力,聚合能力较强。十一元以上环的跨环张力消失,环较稳定,不易聚合。")
101
环烷烃: 在热力学上容易开环程度:3,4>8>7,5 动力学因素:环烷烃极性小,不容易被引发剂活性中所进攻。 实际应用:工业上很少用环烷烃作开环聚合的原料。 杂环:热力学上的开环倾向可近似参照环烷烃的数据 特殊性:杂环的聚合能力还与环中杂原子有关。 杂环中的杂原子提供了引发剂亲核或亲电进攻的位置,因此在动力学上比环烷烃更容易开环聚合。 环上取代基对聚合都不利,因为线性聚合物中取代基间的斥力比环状单体还要大。

102
五元环中的四氢呋喃能够聚合,而γ-丁氧内酯却不能聚合。六元环醚都不能聚合,如四氢吡喃和1,4-二氧六环,但相应的环酯却都能聚合,如环戊内酯。其他六元的环酰胺、环酐都较易聚合。
环上有取代基时对聚合不利。如四氢呋喃能聚合,2-甲基四氢呋喃却不能聚合。

103
2 杂环开环聚合机理和动力学特征 离子型或分子型引发剂,形成离子或中性分子线性活性种,然后继续开环而增长。 引发剂:
2 杂环开环聚合机理和动力学特征 离子型或分子型引发剂,形成离子或中性分子线性活性种,然后继续开环而增长。 引发剂: 离子型引发剂:阳离子引发剂和阴离子引发剂(活泼)。 分子型引发剂:主要是水,但只能引发较活泼的单体。 含氧杂环,因为氧原子易受阳离子进攻,如环氧化合物、环内酯、环酐等可以用阳离子引发剂。 一些含氧三元环合物,张力大,也可用阴离子引发剂引发。环内酰胺一般用碱金属等阴离子引发剂来引发,也可用分子型引发剂来开环。
。 分子型引发剂:主要是水,但只能引发较活泼的单体。 含氧杂环,因为氧原子易受阳离子进攻,如环氧化合物、环内酯、环酐等可以用阳离子引发剂。 一些含氧三元环合物,张力大,也可用阴离子引发剂引发。环内酰胺一般用碱金属等阴离子引发剂来引发,也可用分子型引发剂来开环。")
104
离子开环具有烯类离子聚合的基本特征 : 如溶剂和反离子的影响,由离子对和自由离子活性种来增长,单体只能加到活性种上,活性链之间不能起增长反应。 大部分离子开环聚合属于连锁聚合机理,但有些也带有逐步的性质,可由分子量随时间的变化判断。 自由基聚合:不考虑凝胶效应,分子量与聚合时间或转化率无关,即先后形成的聚合物分子量相近。 阴离子活性聚合:分子量随聚合时间或转化率不断增加,几乎成线性关系。 逐步聚合:大部分聚合时间内或<90%~95%转化率都保持低分子量,直至转化率和高(>98%),才能得到高分子量。 开环聚合可由分子量随时间的变化来判断机理。
,才能得到高分子量。 开环聚合可由分子量随时间的变化来判断机理。")
105
3 元环单体,可进行阴离子、阳离子、配位聚合;
4, 5, 6元环单体,只能阳离子机理。

106
3 环氧烷烃的阴离子开环聚合 环氧烷烃属于三元环,张力大。易受阳离子、阴离子、甚至中性(水)进攻,使C-O键断裂开环。
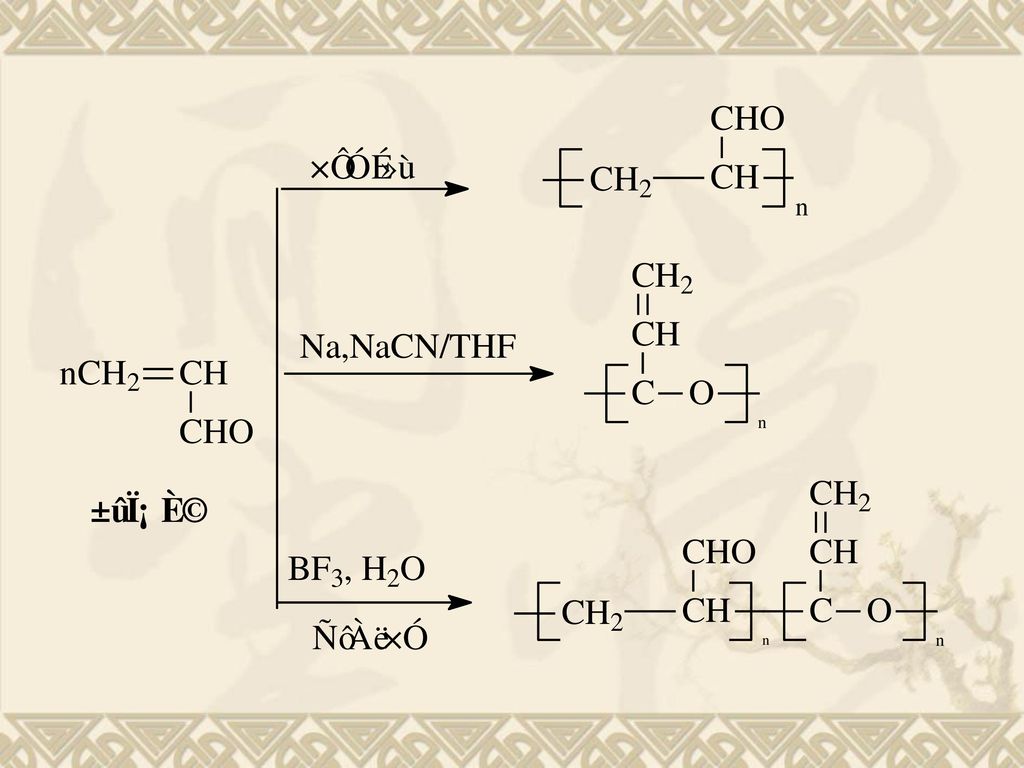
3 环氧烷烃的阴离子开环聚合 环氧烷烃属于三元环,张力大。易受阳离子、阴离子、甚至中性(水)进攻,使C-O键断裂开环。 环上氧原子电子云密度大,易受阳离子进攻,但阳离子开环常伴有链转移反应,因此工业上多采用阴离子引发剂开环聚合。 环氧烷烃中碳原子缺电子,易受阴离子进攻开环。所得分子量可高达百万。
进攻,使C-O键断裂开环。 环上氧原子电子云密度大,易受阳离子进攻,但阳离子开环常伴有链转移反应,因此工业上多采用阴离子引发剂开环聚合。 环氧烷烃中碳原子缺电子,易受阴离子进攻开环。所得分子量可高达百万。")
107
聚醚的结构特征和用途 环氧烷烃开环聚合的产物为聚醚,主要用作非离子型表面活性剂和聚氨酯的预聚物。 非离子型表面活性剂由疏水和亲水两部分组成。 亲水:聚氧乙烯链段; 疏水:由特定起始剂提供,烷基酚等。 环氧乙烷阴离子开环聚合的机理和动力学 引发剂:碱金属的烷氧化物、氢氧化物、氨基化物、有机金属化合物、碱土金属氧化物。

108
以醇钠为引发剂为例,环氧化物开环聚合的机理如下:
必须去除 才能引发开环聚合 以醇钠为引发剂为例,环氧化物开环聚合的机理如下:

109
环氧乙烷的开环聚合属于二级亲核取代反应,速率与环氧乙烷浓度、引发剂浓度成正比,其聚合速率和数均聚合度表达式与阴离子活性聚合完全相同:
其中:[M]0和[M]t分别为环氧乙烷起始和t时刻的浓度;[C]0为引发剂浓度,[C]为 t 时刻的引发剂浓度。

110
交换反应:聚环氧乙烷活性种与起始剂起交换反应,生成起始剂活性种,再引发单体增长。
环氧乙烷活性种与脂肪醇的交换反应: 交换的结果,使原来的活性链终止,导致分子量降低。新形成的起始剂活性种可以再引发单体增长,起始剂就相当于引发剂的作用,因此,聚合度应为: 由于起始剂酸性、引发剂活性的不同,引发、增长、交换反应的相对速率也有差异,最终将使聚合速率、产物分子量各不相同。

111
环氧乙烷的开环聚合-PEO的制备 水溶性高分子,不管分子量多大。 分子量<600, 粘稠液体; 分子量>1000, 蜡状固体。 容易结晶,有熔点。 分子量1000:37-40oC,1500: 43-46oC,20000: 63oC

112
环氧乙烷阴离子开环聚合无链转移反应,分子量3万到4万。
环氧丙烷阴离子开环聚合中的链转移反应 环氧乙烷阴离子开环聚合无链转移反应,分子量3万到4万。 环氧丙烷阴离子开环聚合中,阴离子活性种容易夺取环氧丙烷分子中甲基的氢原子而转移,分子量3000到4000。

113
单体消失速率=增长速率+转移速率 无终止反应,聚合物仅由链转移生成 聚合物生成速率 令CM=ktr,M/kp

114
向单体转移 无转移 开环聚合的CM一般为10-2,比自由基聚合的CM大102~103倍。环氧丙烷聚合中链转移的影响很大,因此一般得不到高分子量聚合物,通常3000~4000(聚合度50~70)左右。
左右。")
115
4 其他环醚的阳离子开环聚合 四、五元环醚的环张力较小,热力学聚合倾向减弱,阴离子不足以进攻极性较弱的碳原子,因此只能用阳离子来进攻极性较强的氧原子,才能进行开环聚合。

116
丁氧环 丁氧环是四元环醚,有较大的环张力,环张力的释放是开环聚合的重要推动力。工业上常用丁氧环的衍生物—3,3’-二(氯亚甲基)丁氧环以三氟化硼等阳离子引发剂,开环聚合成氯化聚醚树脂,用作工程塑料。 三级氧鎓离子,是环醚阳离子开环聚合的活性种 。

117
四氢呋喃 THF是五元环,环张力较小,聚合活性较低。对引发剂和单体的纯度有较高要求。引发剂可用五氯化磷络合物,得到分子量30万的聚四亚甲基氧。 也可用路易斯酸引发,但开环速率较慢,因此常用少量环氧乙烷作为四氢呋喃开环聚合的促进剂,因为路易斯酸络合物所提供的质子很容易引发活性较高的环氧乙烷形成氧鎓离子,然后再由这些氧鎓离子来引发THF开环。

118
环醚活性:环氧乙烷>丁氧环>四氢呋喃>
七元环醚>己氧环(=0) 主要应用:与二异氰酸酯反应生成聚醚聚氨酯。 用做弹力纤维或制备人工心脏材料。
 主要应用:与二异氰酸酯反应生成聚醚聚氨酯。 用做弹力纤维或制备人工心脏材料。")
119
三氧六环(三聚甲醛)的阳离子开环聚合 三氧六环是甲醛的三聚体,易受三氟化硼-水体系引发,进行阳离子开环聚合。
反应机理:引发反应是H+A-与三氧六环形成氧鎓离子,而后开环转化为碳阳离子;碳阳离子成为增长种,增长就是单体插入CH2+···A-之间进行。 H

120
三聚甲醛开环聚合时,存在聚甲醛-甲醛的现象
在聚合结束时,仍存在这种平衡,如果排除甲醛,将打破平衡,使聚甲醛不断解聚。 均聚甲醛受热时,从末端开始,作连锁降解。有两个方法改进。

121
1 聚合结束前,加入醋酐做端基封锁剂,乙酰化,防止聚甲醛从端基开始降解。称为均聚甲醛。
2 与少量二氧五环共聚,在聚甲醛主链中引入-OCH2CH2-链节,使聚甲醛受热降解至此,就不至于继续降解下去,这样的产品成为共聚甲醛。 聚合方法:溶液聚合和本体聚合

122
己内酰胺的阴离子开环聚合 己内酰胺是七元环,存在一定环张力,热力学上有开环局和倾向,最终产物中线性聚合物和环状单体并存,构成平衡。己内酰胺可用酸、碱或水引发开环。阳离子开环聚合无实用价值,分子量和转化率低。 水做引发剂:由己内酰胺合成尼龙-6纤维时,属逐步聚合机理。 碱金属或其衍生物引发剂:属阴离子开环聚合机理。引发后的预聚体可直接浇铸入模内制成铸件,故称为模内浇铸尼龙。

123
引发分为2步:(1)己内酰胺与碱金属M或衍生物B-M+ 反应,形成内酰胺阴离子活性种(I ):
己内酰胺的阴离子开环聚合机理 : 引发分为2步:(1)己内酰胺与碱金属M或衍生物B-M+ 反应,形成内酰胺阴离子活性种(I ): (I) 这是一个平衡反应,必须真空除去副产物BH,使平衡向右移动。然后,内酰胺阴离子与单体反应而开环,生成活泼的胺阴离子(II)。
己内酰胺与碱金属M或衍生物B-M+ 反应,形成内酰胺阴离子活性种(I ): (I) 这是一个平衡反应,必须真空除去副产物BH,使平衡向右移动。然后,内酰胺阴离子与单体反应而开环,生成活泼的胺阴离子(II)。")
124
(2)内酰胺阴离子活性种(I)与另一己内酰胺单体分子反应,形成活泼的胺阴离子活性种(II):
慢 胺阴离子(II)无共轭作用,较活泼,很快夺取另一单体己内酰胺分子上的一个质子,生成二聚体( III ),同时再生内酰胺阴离子(I)。
无共轭作用,较活泼,很快夺取另一单体己内酰胺分子上的一个质子,生成二聚体( III ),同时再生内酰胺阴离子(I)。")
125
(II)
")
126
增长反应首先是活性较高的N—酰化内酰胺与内酰胺阴离子反应,使N—酰化内酰胺开环。
( III ) ( I ) ( 反应4) 反应产物很快再与单体发生质子交换反应,再生出内酰胺阴离子(I)
 ( I ) ( 反应4) 反应产物很快再与单体发生质子交换反应,再生出内酰胺阴离子(I)")
127
( 反应5) 可见,己内酰胺阴离子聚合有两个与其它聚合明显不同的特点:① 活性中心不是自由基、阴离子或阳离子,而是酰化的环酰胺键;② 不是单体加成到活性链上,而是单体阴离子加成到活性链上。
 可见,己内酰胺阴离子聚合有两个与其它聚合明显不同的特点:① 活性中心不是自由基、阴离子或阳离子,而是酰化的环酰胺键;② 不是单体加成到活性链上,而是单体阴离子加成到活性链上。")
128
基于上述特点,己内酰胺的开环聚合反应速率与单体浓度无关,而与活化单体和内酰胺阴离子(I)浓度有关,亦即与引发剂碱性物质的浓度有关,因此速率决定于碱的浓度。
酰化的内酰胺比较活泼,是聚合的活性中心,因此可以采用酰氯、酸酐、异氰酸酯等酰化剂与单体反应,使己内酰胺先形成N-酰化己内酰胺。这样可消除诱导期,加速反应,缩短聚合周期。

129
环硅氧烷的开环聚合 聚硅氧烷是半有机高分子,耐高温、耐化学品。主要产品:硅油,硅橡胶、硅树脂。 氯硅烷水解成硅醇,再脱水缩聚成聚硅氧烷。 -

130
酸水解有利于形成环状或低分子量线性聚合物,碱水解时有利于形成分子量较高的线性聚合物,超高分子量的聚硅氧烷则由环硅氧烷开环聚合而成。
硅氧烷易形成八元环,在经阴离子或阳离子(强质子酸)开环聚合成高分子量的聚硅氧烷,用作硅橡胶。 碱金属的氢氧化物或烷氧化物是环状硅氧烷的常用阴离子引发剂,可使硅氧键断裂,形成硅氧阴离子活性种,环状单体插入 离子键中增长。
开环聚合成高分子量的聚硅氧烷,用作硅橡胶。 碱金属的氢氧化物或烷氧化物是环状硅氧烷的常用阴离子引发剂,可使硅氧键断裂,形成硅氧阴离子活性种,环状单体插入 离子键中增长。")
131
引发 增长 强质子酸或 Lewis 酸也可使硅氧烷开环聚合,活性种是硅阳离子 ,环状单体插入而增长;也可形成氧鎓离子而后重排成硅阳离子。

132
羰基化合物的聚合 羰基化合物中的C=O键经极化后,产生正负电荷两个中心,有异裂倾向,不利于自由基聚合,而适于离子聚合。聚合产物为缩醛。 甲醛结构简单,既可阴离子聚合又可阳离子聚合,是这类化合物的代表。但其精制困难,往往先制成预聚物三聚甲醛,再开环聚合。

133
乙醛以上的高级醛类,由于烷基的位阻效应,聚合热降低。例如乙醛的聚合热仅为29 kJ/mol,因而聚合上限温度很低,仅-31℃,产物分子量很低,无实用价值。同时甲基的诱导效应,使羰基氧上的电子云密度增加,降低了活性种的稳定性,对聚合不利。因此乙醛以上的高级醛类都不能聚合。 丙酮分子上两个甲基导致的位阻效应和诱导效应,使其不能聚合。 醛上的氢被卤素原子取代,由于卤素的吸电子性,使氧上的负电荷密度分散,活性种得到稳定,容易被弱碱引发阴离子聚合。如三氯乙醛、三氟乙醛都容易聚合,甚至可用BPO引发自由基聚合。

134
自由基

136
环状胺和环状硫化物的开环聚合: 只进行阳离子开环聚合,聚合物有支链,水溶性高分子,聚乙烯亚胺。 三元环状硫化物与环氧乙烷、环氧丙烷一样可发生阳离子、阴离子开环聚合反应。

137
N-羧基-a-氨基酸酐(NCA)的开环聚合
碱(如胺,醇盐等)生成聚氨基酸,聚合反应过程中伴随二氧化碳的脱去。 R 不能含活性基团,否则要进行保护。聚合机理比较复杂。多用伯胺引发。
生成聚氨基酸,聚合反应过程中伴随二氧化碳的脱去。 R 不能含活性基团,否则要进行保护。聚合机理比较复杂。多用伯胺引发。")
138
聚对二甲苯




:核蛋白体组成成分 转移 RNA ( tRNA ):转运氨基酸 信使 RNA ( mRNA ):蛋白质合成模板 不均一核 RNA ( hnRNA ):成熟 mRNA 的前体 小核 RNA ( snRNA ):>")










>")




是由30个碳原子组成的萜类化合物。(指基本骨架,不包括糖),可认为是由6个异戊二烯缩合而成的。 分类 从结构上分两大类:四环三萜 五环三萜 存在形式:游离形式(苷元) 苷的形式(与糖结合)>")
