Download presentation
1
心力衰竭的发生机制

2
一种病理生理过程而不是一种疾病 心血管疾病最常见的死亡原因 英国:45~65岁 近20年增加了1.5倍 65岁以上 近20年增加了4倍 美国:每年心衰住院病人100万 我国:脑血管病、恶性肿瘤及心脏病为城 镇居民的前三位死因,占总死亡数的60.3% 预后差:5年生存率仅50%,其恶性程度不低于癌 症及艾滋病

3
调控心排出量的三个变量: 心室充盈量 心肌舒缩活动的强度和速度 心率 循环衰竭的概念 心力衰竭的概念 由于心脏自身的泵血功能严重受损,表现为心排出量减少,不能满足组织的代谢需求,以及神经-体液调节活动异常的病理过程,称为心力衰竭。

4
原观点: 心力衰竭是单纯的心脏工作性能低下引起的血流动力学紊乱 90年代初(1992) Packer MP提出解释心力衰竭进展的神经-内分泌假说,认为心衰是神经-内分泌系统介导的,涉及心、血管、肾、骨骼肌等许多器官、组织的慢性全身性适应反应,其代价是心脏重塑和心功能进行性降低。
 Packer MP提出解释心力衰竭进展的神经-内分泌假说,认为心衰是神经-内分泌系统介导的,涉及心、血管、肾、骨骼肌等许多器官、组织的慢性全身性适应反应,其代价是心脏重塑和心功能进行性降低。 .")
5
第一节 心脏泵血功能损害的适应和代偿机制 心脏对工作负荷增加及神经-体液调节改变的适应 神经-体液调节机制对心泵功能损害所引发的血流 动力学稳态破坏趋势的适应 组织(肾、骨骼肌等)对低灌流状态的适应 代偿 失代偿
对低灌流状态的适应 代偿 失代偿")
6
一、心脏的代偿和适应 迅速启动的代偿机制(功能性调整) 缓慢持久的适应机制(结构性改建)
 缓慢持久的适应机制(结构性改建)")
7
心室负荷过重 神经-体液调节 机制激活 心肌肥大 静脉回心血量↑ 心肌收缩能力↑ 心输出量↑ 搏出量↑ 心率↑

8
1、动用心功能贮备 (1) 增加前负荷,通过异长调节(Starling机制)使搏出量增加。 神经-体液调节机制 钠水潴留 容量血管收缩 静脉回心血量 心室舒张末容量(充盈压) LV充盈压:5~ ~15mmHg 肌小节:1.7~ ~2.2um 心肌细胞收缩强度 ,搏出量
 增加前负荷,通过异长调节(Starling机制)使搏出量增加。 神经-体液调节机制 钠水潴留 容量血管收缩 静脉回心血量 心室舒张末容量(充盈压) LV充盈压:5~6 12~15mmHg 肌小节:1.7~ ~2.2um 心肌细胞收缩强度 ,搏出量")
9
利: 弊: 左室舒张末容量只能增加10%,代偿有限 ①充盈压的过度增加使静脉淤血加重; 心肌固有的自身调节机制,快速、应急性调节
②心腔半径增大使收缩期室壁应力增加,导致心肌耗氧量增大; ③舒张末压升高和心肌静息张力增大,增加了心脏舒张期血液灌注阻力,可致心内膜下心肌缺血。

10
(2) 心肌收缩能力增强,通过等长调节使搏出量增加 SNS EP、NE -R
胞浆cAMP PKA 钙通道蛋白磷酸化 [Ca2+]i 升高速率和幅度 急性期,可维持心输出量和血流动力学稳态 慢性期,心肌收缩力降低及心肌对儿茶酚胺的反应性降低,意义不大

11
(3) 心率加快 利:维持心输出量 弊: HR 150 bpm SNS 正性变时,正性变传导 HR HR 170~180 bpm
心肌耗氧量 ; 冠脉血流 临床应用: -阻滞剂,洋地黄

12
2、心肌肥大与心室重构 由于心肌细胞、非心肌细胞及细胞外基质在基因表达改变的基础上所发生的变化,使心脏的结构、代谢和功能都经历了一个模式改建的过程,称为心室重构(ventricular remodeling),或心肌改建(myocardial remodeling)。 心肌肥大 心肌细胞表型改变 非心肌细胞及细胞外基质改建
,或心肌改建(myocardial remodeling)。 心肌肥大 心肌细胞表型改变 非心肌细胞及细胞外基质改建")
13
(1) 心肌肥大 肥大:功能负荷增加导致器官大小的比值增大
心肌细胞体积增大伴非心肌细胞及细胞外基质相应增多所致的心室重量或(和)室壁厚度增加。 在细胞水平 心肌细胞体积增大(myocyte hypertrophy) 在组织水平 心肌质量增加(myocardial hypertrophy) 压力超负荷性心肌肥大(向心性) 容量超负荷性心肌肥大(离心性)
室壁厚度增加。 在细胞水平 心肌细胞体积增大(myocyte hypertrophy) 在组织水平 心肌质量增加(myocardial hypertrophy) 压力超负荷性心肌肥大(向心性) 容量超负荷性心肌肥大(离心性)")
14
利: 弊: 适应室壁应力的变化并最终使室壁应力“正常化”。 Laplace定律,S = pr/2h
向心性肥大可致心肌缺血及舒张功能异常,而离心性肥大可致功能性二(三)尖瓣返流及收缩功能异常; 由于心肌细胞表型改变及间质胶原增生,肥大心肌最终会由于继发的舒缩功能降低而走向衰竭,两类心肌肥大都会转向进行性心腔扩大。
尖瓣返流及收缩功能异常; 由于心肌细胞表型改变及间质胶原增生,肥大心肌最终会由于继发的舒缩功能降低而走向衰竭,两类心肌肥大都会转向进行性心腔扩大。")
15
(2) 心肌细胞表型(phenotype)改变 由于所合成的蛋白质的种类变化所致的心肌细胞“质”的改变
主要机制:同工型转换(isoform switches) 正常基因的表达改变(失活或活化) 胎儿期基因被激活 某些基因表达受到压制 某些基因表达过度、缺失或突变 如:线粒体基因(mtDNA)与核基因(nDNA)
 正常基因的表达改变(失活或活化) 胎儿期基因被激活. 某些基因表达受到压制. 某些基因表达过度、缺失或突变. 如:线粒体基因(mtDNA)与核基因(nDNA)")
16
成分 变化 mRNA水平 蛋白质水平 α心肌肌球蛋白重链 β心肌肌球蛋白重链 肌球蛋白轻链-1 胚胎/心房型 心室型 利钠多肽
β肾上腺素受体 Ⅰ型胶原前胶原蛋白 Ⅲ型胶原前胶原蛋白 ……………. ↓ ↑ +

17
意义: 正常功能改变 通过分泌的细胞因子和局部激素而相互作用,进一步促进细胞生长、增殖及表型改变,从而使细胞器发生了在蛋白质水平的变化。
新近(1999)发现有些基因突变对机体是有利的,如:一磷酸腺苷脱氨酶-I(AMPD-I)基因突变
发现有些基因突变对机体是有利的,如:一磷酸腺苷脱氨酶-I(AMPD-I)基因突变.")
18
(3)非心肌细胞增生及细胞外基质改建 非心肌细胞(占细胞总数的2/3 ): 成纤维细胞、血管平滑肌细胞、内皮细胞 细胞外基质(ECM):
结构糖蛋白、蛋白多糖和糖胺聚糖 最主要的是纤维状的I型和Ⅲ型胶原

19
表达α-SMA和粘附分子使其能迁移、收缩; 表达组织蛋白酶D参与局部RAS激活 分泌大量胶原及ECM其它成分,调控胶原酶
机械负荷 多潜能间质成纤维细胞 心脏成纤维细胞 化学信号 表达生长因子及其受体,促增殖 表达α-SMA和粘附分子使其能迁移、收缩; 表达组织蛋白酶D参与局部RAS激活 分泌大量胶原及ECM其它成分,调控胶原酶 的活性,促使胶原网络的生化改建及结构改建

20
利: 弊: 心肌的僵硬度增大, 早期 Ⅲ型胶原 侧向连接 伸展性及回弹性较好,对于心肌细胞肥大及肌束组合的重新排列十分有利
早期 Ⅲ型胶原 侧向连接 伸展性及回弹性较好,对于心肌细胞肥大及肌束组合的重新排列十分有利 后期 I型胶原 与心肌束平行排列 可提高心肌的抗张强度,防止在室壁应力过高的情况下,心肌细胞侧向滑动而造成的室壁变薄和心腔扩大。 弊: 心肌的僵硬度增大, 心肌收缩的内阻力增大 妨碍血管扩张和血流量增加

21
(4)心肌改建的细胞和分子机制 ①刺激心肌肥大和改建的信号及其整合 机械信号 刺激信号 化学信号 代谢信号 ②跨膜信号传递: 化学信号 受体 蛋白激酶(PKC) 机械信号 刺激生长因子释放 激活应力感受器 细胞骨架
心肌改建的细胞和分子机制 ①刺激心肌肥大和改建的信号及其整合 机械信号 刺激信号 化学信号 代谢信号 ②跨膜信号传递: 化学信号 受体 蛋白激酶(PKC) 机械信号 刺激生长因子释放 激活应力感受器 细胞骨架")
22
③即刻早期反应基因(immediate early gene)激活:
30分钟内作为原癌基因的即刻早期基因(如c-fos,c-myc,c-jun,egr1及c-ras)激活; 6~12小时,胎儿期表达的基因重新激活(如β-MHC),而相应的成年心脏表达的一些基因部分失活(如α-MHC),导致同工型转换; 12~24小时,无同工型转换的固有基因上调(如肌球蛋白轻链-2、α心肌肌动蛋白); 24小时后,心肌细胞内蛋白质及RNA总量增加,细胞体积开始增大。
激活; 6~12小时,胎儿期表达的基因重新激活(如β-MHC),而相应的成年心脏表达的一些基因部分失活(如α-MHC),导致同工型转换; 12~24小时,无同工型转换的固有基因上调(如肌球蛋白轻链-2、α心肌肌动蛋白); 24小时后,心肌细胞内蛋白质及RNA总量增加,细胞体积开始增大。")
23
牵拉和血管紧张素Ⅱ所引起的心肌细胞肥大性反应的比较
AngⅡ 蛋白质合成 蛋白质含量 DNA合成 即刻早期基因表达(c-fos等) 胎儿型基因表达(ANF等) 生长因子基因表达(TGF-β等) AT1受体阻滞剂的阻断作用 ↑ → 有
 胎儿型基因表达(ANF等) 生长因子基因表达(TGF-β等) AT1受体阻滞剂的阻断作用. ↑ → 有.")
24
最近研究表明:心肌重塑可由基因突变启动 如过度表达一种肌浆网钙结合蛋白(钙隐蛋白)的小鼠会发生适应性左室肥厚;而过度表达人β2肾上腺素受体的小鼠,其心肌收缩力显著增强,不发生左室肥厚,舒张性能增强。 心肌重塑可由内在(基因异常)和外部(调节信号)两类因素启动。
和外部(调节信号)两类因素启动。")
25
改建刺激 机械性 神经体液性 细胞因子 血管紧张素 儿茶酚胺 内皮素 多肽生长因子 炎症细胞因子 NO 非心肌细胞 心肌细胞 心肌改建 心肌肥大 表型改变 细胞外基质改变 功能改变

26
传统的观点认为,心肌细胞是终末分化细胞,不进入细胞周期。
3、心肌细胞生长 传统的观点认为,心肌细胞是终末分化细胞,不进入细胞周期。 最近的研究结果证实,有15%~20%的心肌细胞保留有复制分裂的能力。

27
(二)神经-内分泌系统激活 1、交感神经系统活性增强及血浆儿茶酚胺浓度升高 机制: 抑制性传人信号减弱和兴奋性传人信号增强 压力感受器、心肺感受器 化学感受器 代谢感受器
神经-内分泌系统激活 1、交感神经系统活性增强及血浆儿茶酚胺浓度升高 机制: 抑制性传人信号减弱和兴奋性传人信号增强 压力感受器、心肺感受器 化学感受器 代谢感受器")
28
利: 心肌收缩力 ,心率 RAS激活,肾血管收缩,血管加压素释放 钠水潴留 回心血量 阻力血管收缩,维持血压并保证重要器官的灌注

29
①使心室的前、后负荷增重及心肌耗氧量增大;
失代偿: ①使心室的前、后负荷增重及心肌耗氧量增大; ②通过α1肾上腺素受体和β肾上腺素受体介导的直接作用,使心肌细胞及成纤维细胞表型改变,促进心室重构; ③诱发心律失常; ④通过使心肌细胞内Ca2+过多及自由基产生增多而对心肌细胞发挥直接的毒性作用。

30
2、肾素-血管紧张素系统激活 激活途径: 心输出量 ①交感神经兴奋,刺激肾小球旁器的β1肾上腺素能受体;
②肾血流量减少激活肾血管的压力感受器; ③滤过率降低及交感神经兴奋所致的近端小管重吸收率增高使致密斑的钠负荷降低。

31
意义: 缩血管,刺激醛固酮的释放并增强交感神经释放NE,因此可使心室的前、后负荷均明显增加 抑制组织RAS并使激肽系统活性升高
AngⅡ可通过心肌细胞及成纤维细胞的血管紧张素受体(主要是AT1受体),刺激心肌细胞肥大和成纤维细胞增殖并使两者的表型改变
,刺激心肌细胞肥大和成纤维细胞增殖并使两者的表型改变.")
32
(三)外周组织对低灌注的适应 包括血容量增加、血流重分布、红细胞增多、组织细胞利用氧的能力增强等。
外周组织对低灌注的适应 包括血容量增加、血流重分布、红细胞增多、组织细胞利用氧的能力增强等。")
33
综上所述, ①心泵功能损害启动两种主要的适应机制:心室重构以适应工作负荷,神经-激素系统激活以维持器官血流灌注; ②适应本身有两面性:适应和适应不良; ③适应发生于心力衰竭的全过程,适应的两面性推动心力衰竭发展过程,并使其表现出阶段性(代偿→衰竭)。
。")
34
第二节 心肌收缩和舒张性能降低的 细胞和分子机制
第二节 心肌收缩和舒张性能降低的 细胞和分子机制

35
(一)心肌细胞数量减少及结构改变 1、心肌细胞数量减少 (1)心肌细胞坏死 ①缺血性死亡
一、心肌收缩能力降低的机制 (一)心肌细胞数量减少及结构改变 1、心肌细胞数量减少 (1)心肌细胞坏死 ①缺血性死亡 前、后负荷增大使心肌耗氧量增多 肥厚的心室使冠脉血流储备减少 心率加快又进一步使耗氧量增大而舒张期缩短
心肌细胞数量减少及结构改变 1、心肌细胞数量减少 (1)心肌细胞坏死 ①缺血性死亡. 前、后负荷增大使心肌耗氧量增多. 肥厚的心室使冠脉血流储备减少. 心率加快又进一步使耗氧量增大而舒张期缩短.")
36
心肌细胞周围纤维化 体液因子对心肌细胞的直接毒性作用, ②窒息性死亡 舒缩活动的阻力增大; 妨碍与组织液间的物质交换,致心肌 细胞萎缩和死亡
③中毒性死亡 体液因子对心肌细胞的直接毒性作用, 如NE、AngⅡ

38
(2)心肌细胞凋亡 在一定基因调控下的细胞主动性死亡过程
机制:不清,可能与缺氧、TNF、NO的作用及心肌死亡受体Fas表达增强有关。

39
在代偿期,细胞凋亡可致心肌肥厚与后负荷不匹配,使室壁应力增大并进一步刺激重构与凋亡。
意义: 在代偿期,细胞凋亡可致心肌肥厚与后负荷不匹配,使室壁应力增大并进一步刺激重构与凋亡。 在衰竭期,心肌细胞凋亡及坏死又可致室壁变薄,心室进行性扩大。 临床:干预凋亡的治疗,如应用生长激素治疗扩张性心肌病伴心衰病人。

40
中期:肌丝不成比例增加,肌节不规则叠加、肌原
2、心肌细胞和心肌组织的结构改变 初期:线粒体数目增多、体积增大 肌原纤维增多 细胞核增大 中期:肌丝不成比例增加,肌节不规则叠加、肌原 纤维排列紊乱; 细胞骨架中的微管密度增大并平行于肌原纤 维排列,使心肌细胞内肌丝滑行的阻力增大, 细胞的缩短速率减慢(-45%)
")
41
晚期:肌原纤维减少; 部分心肌细胞萎缩; 细胞内各种细胞器的比例失衡; 心肌细胞之间发生侧向移动与错位

42
3、心室扩张 机制: 心肌细胞数减少; 弊:前负荷增大 细胞骨架改变引起肌丝重排,心肌细胞体积不变而长度增大;
室壁应力增大可使基质金属蛋白酶的表达和活性均增高,胶原降解增强,导致心肌细胞之间发生侧向滑动与错位。 弊:前负荷增大 功能性二尖瓣返流

43
(二)能量代谢障碍 1、能量产生及贮存减少 (1)心肌细胞缺血 冠脉血流储备减少 心肌细胞肥大性生长 (2)心肌细胞能量产生障碍 肌原纤维不成比例的增加使线粒体数目相对减少; 线粒体利用氧的能力降低
能量代谢障碍 1、能量产生及贮存减少 (1)心肌细胞缺血 冠脉血流储备减少 心肌细胞肥大性生长 (2)心肌细胞能量产生障碍 肌原纤维不成比例的增加使线粒体数目相对减少; 线粒体利用氧的能力降低")
44
(3)心肌贮能减少 心肌主要贮能形式:CP 心衰时ATP、CP及肌酸含量均减少, CP/ATP降低 (4)收缩蛋白ATP酶活性降低
与心肌调节蛋白改变有关。例如肌球蛋白轻链-1及肌钙蛋白T亚单位的胎儿型同工型增多,以及肌钙蛋白I亚单位的磷酸化状态减低等。

45
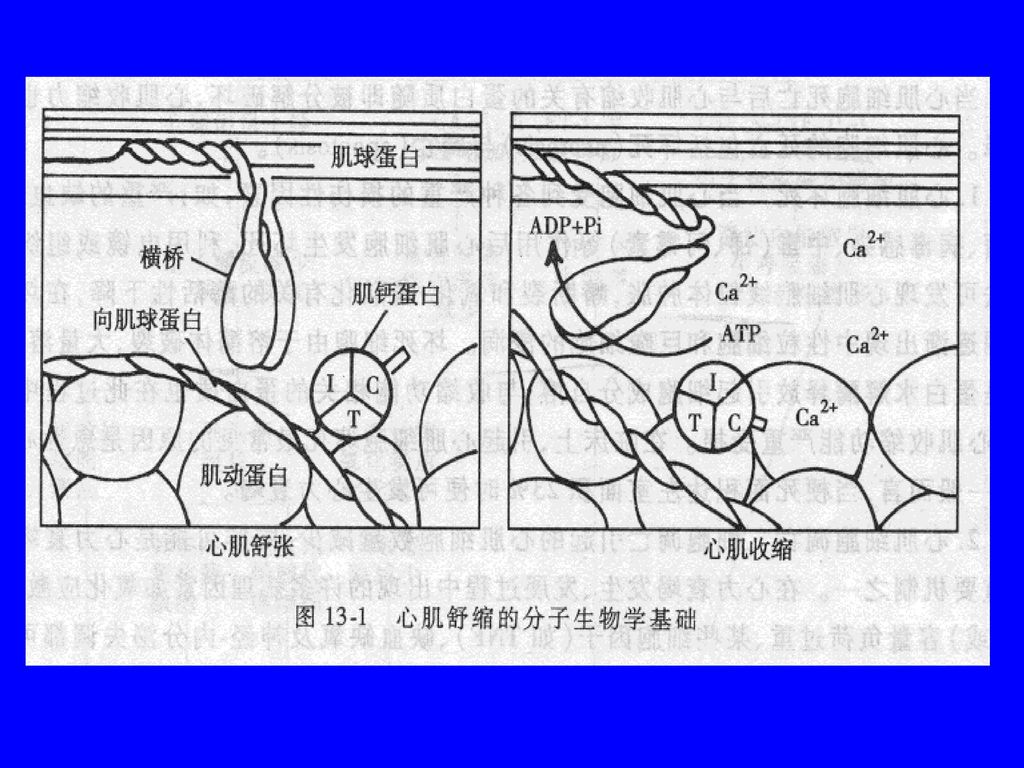
(三) 兴奋-收缩耦联障碍 1、Ca2+内流减慢及SR摄取、释放Ca2+能力降低 SR钙ATP酶减少 SR摄钙、贮钙减少 SL电压门控钙通道、SR钙释放通道减少 或两者间的耦联障碍 心肌兴奋时钙内流和SR钙释放减少 兴奋时Ca2+ 瞬变峰值降低,从而使活化横桥数 减少而致心肌收缩能力降低
 兴奋-收缩耦联障碍 1、Ca2+内流减慢及SR摄取、释放Ca2+能力降低 SR钙ATP酶减少 SR摄钙、贮钙减少 SL电压门控钙通道、SR钙释放通道减少 或两者间的耦联障碍 心肌兴奋时钙内流和SR钙释放减少 兴奋时Ca2+ 瞬变峰值降低,从而使活化横桥数 减少而致心肌收缩能力降低")
46
2、心肌对儿茶酚胺正性变力作用的反应性降低
NE耗竭、合成减少 NE含量 1 -R下调、2 -R部分脱耦联 Gs-蛋白减少,Gi-蛋白增多,使Gi/Gs

47
(二)心肌舒张性能异常的机制 1、心肌的主动松弛异常 2、心肌顺应性 顺应性:可伸展性(dv/dp) 心肌缺血 ATP
SR摄钙速率及SL向胞外转运钙的速率 心肌肥大 2、心肌顺应性 顺应性:可伸展性(dv/dp) 心肌质量及室壁厚度增大 心肌的固有僵硬度增大
 心肌质量及室壁厚度增大. 心肌的固有僵硬度增大.")
48
原 发 病 心室泵血功能障碍 心室重构 神经-体液调节 机制激活 心力衰竭综合征 低灌流综合征 静脉淤血综合征 病残 死亡 诱 因 血管收缩 钠水潴留 血流动力学 稳态破坏

49
第三节 目前研究的几个热点

50
一、细胞因子与心力衰竭 (一) 白细胞介素-1家族 (二)白细胞介素-6家族 (三)肿瘤坏死因子(TNF)
分为TNF-α和TNF-β,来源于巨噬细胞、激活的淋巴细胞、NK细胞、LAK细胞、内皮细胞和血管平滑肌细胞。 TNF以及IL-1、IL-6的作用与NO有关。它们通过激活心肌细胞膜上及胞内诱导型NOS(iNOS),使NO水平显著升高,从而导致心肌收缩力下降
,使NO水平显著升高,从而导致心肌收缩力下降.")
51
(四)生长因子系列 5个家族:EGFS、FGFS、IGFS、PDGFS 和TGFS 除EGFS外,其余4类生长因子均可引起心肌细胞肥大
生长因子系列 5个家族:EGFS、FGFS、IGFS、PDGFS 和TGFS 除EGFS外,其余4类生长因子均可引起心肌细胞肥大")
52
(五)核因子-kappa B NF-κB是一种核蛋白因子,能与免疫球蛋白κ轻链基因增强子κB序列特异性结合 1、心肌炎性反应与心力衰竭
NF-κB亚单位IκB磷酸化和降解使NF-κB核易位是急性炎症发生发展的始动机制。TNF-α可刺激心肌细胞膜IκB降解,进而激活NF-κB,导致依赖NF-κB激活的基因转录并与核DNA结合增加以诱导心肌细胞凋亡,从而引起或加重心衰。

53
2、NF-κB与心肌细胞凋亡 血管紧张素Ⅱ可通过激活单个核细胞NF-κB,进一步促进前炎性细胞因子的分泌而参与炎症过程的调控。已知的动物模型中,TNF-α的生物活性由Fas介导,Fas是调控细胞凋亡信号途径的重要基因。因此上述细胞因子的相互作用,推测可能是导致心衰的部分机制之一。

54
3、NF-κB与心室重构 TNF-α激发心肌细胞肥大生长反应,可能对心肌外源损伤后的稳定起重要作用,NF-κB的激活可能是心肌肥大的中间机制之一。

55
二、NO与心力衰竭 来源于血管内皮上L-精氨酸的氨基端,在NOS 的作用下被合成
三种一氧化氮合成酶(NOS)的亚型:n NOS、i NOS和e NOS
的亚型:n NOS、i NOS和e NOS.")
56
心衰患者血浆内的NO含量明显升高,尤以心功能Ⅲ、Ⅳ级患者增加明显
机制: ①心衰时TNF-α等细胞因子显著升高,促进NO的释放; ②心衰时神经内分泌过度激活致NO释放增加; ③大量iNOS 激活致NO释放剧增。

57
意义: 对调节外周血管及心脏舒缩功能起一定的代偿作用,但随着心衰病情的发展,NO 释放过量增加,又对心脏具有负性肌力作用,表现在:
可与不同形式的铁反应。过量NO产生时,尤其在过氧化物丰富的环境中,NO可引起细胞内铁明显丢失,结果造成细胞生长和增殖受影响,形成许多具有细胞毒性的活化形式,介导细胞毒性作用,从而诱导心肌细胞的凋亡。

58
NO过多对蛋白质功能的影响 可直接与巯基化物反应形成硝基硫醇(RSNO),参与NO的细胞毒性作用。 参与调节受体蛋白质的功能,使受体的巯基亚硝基化,从而使受体失活,这些修饰均可能经引起酶的抑制,下调葡萄糖水解和能量合成,最终引起心肌细胞死亡以及抑制心肌的收缩力。
,参与NO的细胞毒性作用。 参与调节受体蛋白质的功能,使受体的巯基亚硝基化,从而使受体失活,这些修饰均可能经引起酶的抑制,下调葡萄糖水解和能量合成,最终引起心肌细胞死亡以及抑制心肌的收缩力。")
59
NO过量对能量代谢的影响 抑制三羧酸循环的关键酶 抑制线粒体氧化作用和无氧酵解,减少ATP的合成。

60
NO过多对基因毒性作用 过多的NO可形成有致癌作用的亚硝胺,亚硝胺是DNA的一种烷化因子,而且NO还有抑制机体对烷化DNA的修复作用。 在O2参与下体外NO可加速碱基的脱氨基,引起心肌细胞的基因突变,而且NO本身就可使腺嘌呤、鸟嘌呤和各种DNA、RNA脱氨基。 NO和其产物ONOO-、OH-均可氧化DNA,最终引起DNA链断裂、交叉连接、碱基修饰等各种类型的DNA损害,从而引起心肌细胞发生死亡。

61
NO的负性肌力作用 在心肌和心内膜中,各种因素诱导产生过量NO可抑制心肌的收缩性。如TNF-α、IL-6、IL-2 可剂量依赖性产生对肌肉收缩的抑制作用

62
三、内皮素与心力衰竭 充血性心衰的病人和动物血浆内皮素水平均升高。 机制:生成增多或清除不足。 意义:
早期:内皮素收缩静脉增加心脏前负荷,利于提高心输出量;增加体循环血管阻力,可维持组织灌注压;保水保钠则可维持一定血浆量, 后期:主要起损伤作用,可加重心输出量降低、钠水潴留和外周血管收缩。

63
参考文献 1、病理生理学(七年制规划教材)。陈主初主编,人民卫生出版社,2001,8 2、心血管病理生理学。吴立玲主编,北京医科大学出版社,2000,3 3、Anversa P, Nadal-Ginard B. Myocyte renewal and ventricular remodeling. Nature, 2002, 415: 4、心力衰竭分子机制研究的新进展,陈兰英、姜立群,中国循环杂志, 2002 Vol.17 No.6 5、Fas/FasL与充血性心力衰竭,李源、王晓明、李慧芳,国外医学(老年医学分册), 2002 Vol.23 No.6 6、细胞骨架在充血性心力衰竭中的研究进展,崔华、常丽,国外医学(儿科学分册), 2002 Vol.29 No.6 7、核因子-kappa B与心力衰竭的关系,李培杰、陈天铎、杨兰、马莉,中国循环杂志, 2002 Vol.17 No.4 8、细胞骨架与心力衰竭,迟路湘、陈红勤,中国循环杂志, 2002 Vol.17 No.4 9、粘附分子与充血性心力衰竭,祝善俊、李振魁,免疫学杂志, 2002 Vol.18 No.z1 10、一氧化氮与充血性心力衰竭的研究进展,周亚峰、程绪杰、刘志华,中国血液流变学杂志, 2002 Vol.12 No.2
, 2002 Vol.23 No.6. 6、细胞骨架在充血性心力衰竭中的研究进展,崔华、常丽,国外医学(儿科学分册), 2002 Vol.29 No.6. 7、核因子-kappa B与心力衰竭的关系,李培杰、陈天铎、杨兰、马莉,中国循环杂志, 2002 Vol.17 No.4. 8、细胞骨架与心力衰竭,迟路湘、陈红勤,中国循环杂志, 2002 Vol.17 No.4. 9、粘附分子与充血性心力衰竭,祝善俊、李振魁,免疫学杂志, 2002 Vol.18 No.z1. 10、一氧化氮与充血性心力衰竭的研究进展,周亚峰、程绪杰、刘志华,中国血液流变学杂志, 2002 Vol.12 No.2.")

. 免疫缺陷病 (Immunodeficiency diseade,IDD) : 由免疫系统中任何一个成分在发生、发 育和成熟过程中的缺失或功能不全而导致免 疫功能障碍所引起的疾病。 免疫缺陷病分为 : 先天性 />")

微循环的组成和血流通路微循环的组成和血流通路 1 、组成:七个部分.>")
 心功能不全 华中科技大学同济医学院病理生理系. 病史:患风湿性心脏病 10 余年。近 3 月来出现心慌、 闷气, 伴浮肿、腹胀,不能平卧。 体查:重病容, 唇甲紫绀, 半坐卧位, 颈静脉怒张。 呼 吸 36 次 / 分, 两肺底可闻湿性罗音。心界向左右两侧扩.>")

:核蛋白体组成成分 转移 RNA ( tRNA ):转运氨基酸 信使 RNA ( mRNA ):蛋白质合成模板 不均一核 RNA ( hnRNA ):成熟 mRNA 的前体 小核 RNA ( snRNA ):>")











.>")

>")