Download presentation
1
第六章 分子的结构与性质

2
原子结构: 金属性、非金属性、递变规律 但 同素异性、同分异构 键长 分子结构 空间构型 键角 化学键 离子键 共价键 金属键 分子间作用力(范德华力),氢键
,氢键")
3
分子的结构与性质 键参数 价键理论 分子间力和氢键 分子的几何构型 分子轨道理论

4
6.1 键参数 凡能表征化学键性质的物理量都称为键参数(键极性、强度、空间性质) 一、键能(Eθ)
旧键断裂,新键形成,都会引起体系内能的变化 △U =△H-P△V △H 键能: 气体分子每断裂单位物质的量的某键时的焓变. H2(g)2H(g)(键离解能D=436kJ/mol) CH4(g)C(g)+4H(g) (四级键离解能的平均值) 作用: 衡量化学键牢固程度的键参数
2H(g)(键离解能D=436kJ/mol) CH4(g)C(g)+4H(g) (四级键离解能的平均值) 作用: 衡量化学键牢固程度的键参数.")
5
二、键长(Lb) 键长Lb: 分子内成键两原子核间的平衡距离 同一种键在不同分子中的键长数值上基本上是个定值: 一个键的性质主要决定于成键原子的本性。 两个确定的原子间,如形成不同的化学键,其Lb越短,键能Eθ越大,键越牢固

6
两个相同原子组成的共价单键键长的一半,即共价半径
不同原子组成的键的键长,A-B键的键长,Lb(A-B)=γA+γB(共价半径之和) 测量方法: 分子光谱,X射线衍射
=γA+γB(共价半径之和) 测量方法: 分子光谱,X射线衍射.")
7
三、键角 分子中两个相邻化学键间的夹角称键角 P155图 测试: 分子光谱或X射线衍射法 分子几何构型: 分子内全部化学键的键长和键角数据

8
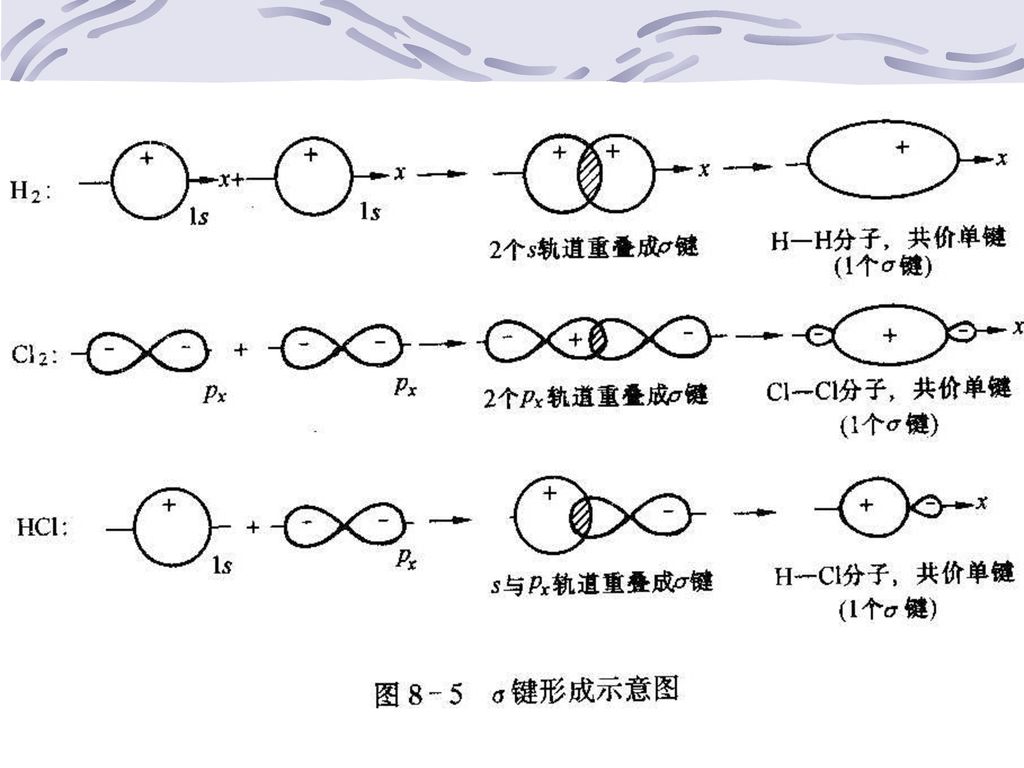
价键理论 现代共价键理论:价键理论和分子轨道理论 二、价键理论 (一)共价键 1、共价键的形成
H2分子核间距=74pm,H原子的波耳半径为53pm (1)在H2分子中两个H原子的1s轨道发生了重叠 两核间形成了一个电子出现的概率密度较大的区域 (2)削弱了两核间的正电排斥力 (3)增强了核间电子云对两氢核的吸引力 体系能量降低 形成共价键
在H2分子中两个H原子的1s轨道发生了重叠. 两核间形成了一个电子出现的概率密度较大的区域. (2)削弱了两核间的正电排斥力. (3)增强了核间电子云对两氢核的吸引力. 体系能量降低. 形成共价键.")
9
共价键——原子间成键电子的原子轨道重叠而形成的化学键。
2、价键理论要点 (1)两原子接近时,自旋方向相反的未成对的价电子可以配对,形成共价键 (2)成键电子的原子轨道如能重叠越多,形成的共价键越牢固——最大重叠原理
两原子接近时,自旋方向相反的未成对的价电子可以配对,形成共价键. (2)成键电子的原子轨道如能重叠越多,形成的共价键越牢固——最大重叠原理.")
10
3、共价键的特征 (1)饱和性 一个原子有几个未成对电子,一般就只能和几个自旋方向相反的电子配对成键,如N≡N,说明一个原子形成共价键的能力是有限的,这就决定了共价键具有饱和性。 有些原子中本来为未成对电子,在特定条件(①原子外层有空轨道,②相化合的原子必须电负性大)下也可被拆开为单电子而参与成键,如SF6的形成。此为激发成键 (2)方向性 成键电子的原子轨道只有沿轨道伸展方向进行重叠(s轨道除外),才会有最大重叠,所以有方向性,如H2O分子。
下也可被拆开为单电子而参与成键,如SF6的形成。此为激发成键. (2)方向性. 成键电子的原子轨道只有沿轨道伸展方向进行重叠(s轨道除外),才会有最大重叠,所以有方向性,如H2O分子。")
11
4、原子轨道的重叠 对称性原则: 只有当原子轨道对称性相同(原子轨道中“+”“-”符号表明对称性)的部分重叠,两原子间才会有概率密度(电子云)较大的区域,才能形成共价键 (原子轨道角度分布图中“+”“-”表示此图形的对称关系:符号相同,表示对称性相同;符号相反,表示对称性不同或反对称)
 .")
12
图6.3

13
5 共价键类型 根据重叠方式不同,可分为σ键(头碰头式), π键(肩并肩式)和键(d轨道面对面) 。 极性共价键 强极性键
极性共价键 强极性键 共价键 弱极性键 非极性共价键 根据重叠方式不同,可分为σ键(头碰头式), π键(肩并肩式)和键(d轨道面对面) 。
, π键(肩并肩式)和键(d轨道面对面) 。")
14
(1) σ键 特点 头碰头式 重叠部分对键轴(两原子核间连线)具有圆柱型对称 绕键轴旋转,形状与符号都不会改变 σ电子
 σ键 特点 头碰头式 重叠部分对键轴(两原子核间连线)具有圆柱型对称 绕键轴旋转,形状与符号都不会改变 σ电子")
16
(2) π键 特点: 肩并肩式 重叠部分对键轴所在的某一特定平面具有反对称性 重叠部分处在该平面的上,下两侧,形状相同但符号相反
π键键能较σ键小。 π 电子 d-d轨道

17
px-px:s键 py-py:p键 pz-pz:p键
如果原子之间只有1对电子,形成的共价键是单键,通常总是s键;如果原子间的共价键是双键,由一个s键一个p键组成;如果是叁键,则由一个s键和两个p键组成。 px-px:s键 py-py:p键 pz-pz:p键

18
(2)π键:原子轨道重叠部分,对键轴所在的某一特定平面具有反对称性,称π键,形成这种键的电子叫π电子。
形成双键或叁键的两原子间,常既有σ键又有π键。如N2分子中。

19
比较s键和p键 s-s: 键,如:H-H s-p: 键,如:H-Cl p-p: 键,如:Cl-Cl 单键: 键 双键:一个 键,一个 键
单键: 键 双键:一个 键,一个 键 叁键:一个 键,两个 键 σ键的重叠程度比π键大,∴π键不如σ键牢固。

20
σ键 π键 原子轨道重叠方式 头碰头 肩并肩 能单独存在 不能单独存在 沿轴转180O 符号不变 符号变 牢固程度 牢固 差 重叠程度 大 小 含共价双键和叁键的化合物的重键容易打开,参与反应

21
(3) 键 一个原子的轨道与另一个原子相匹配的轨道以“面对面”的方式重叠形成的键 dxy与dxy 在金属原子间成键或多核配合物结构中出现
 键 一个原子的轨道与另一个原子相匹配的轨道以 面对面 的方式重叠形成的键 dxy与dxy 在金属原子间成键或多核配合物结构中出现")
22
6、配位共价键 配位键: 凡共用电子对由一个原子单方面提供而形成的共价键 CO

23
配位键形成的条件有二: (1)一个原子其价层有未共用的电子对(孤电子对)。 (2)在原子价层有空轨道。 在分子间、分子内、离子间或分子-离子间均可形成。
一个原子其价层有未共用的电子对(孤电子对)。 (2)在原子价层有空轨道。 在分子间、分子内、离子间或分子-离子间均可形成。")
24
价键理论 (二)离子键 离子键本质——阳、阴离子间静电引力而形成的化学键。离子键可存在于气体分子(如Na+、Cl-离子型分子内),但大量存在于离子晶体中。 特征—无饱和性、无方向性
离子键 离子键本质——阳、阴离子间静电引力而形成的化学键。离子键可存在于气体分子(如Na+、Cl-离子型分子内),但大量存在于离子晶体中。 特征—无饱和性、无方向性")
25
成键两元素的电负性差值(△X)越大,键极性越强
非极性键——极性键——离子键 离子键是强极性键的极限 极性共价键:小部分离子键成分和大部分共价键成分 当极性键向离子键过渡时,共价键部分(又称共价性)渐少,离子性渐增。
渐少,离子性渐增。")
26
6.3 分子的几何构型 (一)价键理论的局限性。 用价键理论能较好地说明不少双原子分子价键的形成,但不能很好说明多原子分子价键的形成和分子构型,如CH4就遇到困难 (二)杂化轨道理论 鲍林在价键理论中引入杂化轨道概念,并发展成为杂化轨道理论
杂化轨道理论. 鲍林在价键理论中引入杂化轨道概念,并发展成为杂化轨道理论.")
27
(2)同一原子中能级相近的n个原子轨道,组合成n个杂化轨道,如sp杂化。 方法:两个轨道相加、相减 结果:
1、杂化轨道理论的要点: (1)某原子成键时,在键合原子的作用下,原来原子的运动状态有可能发生变化,主要有两个方面①电子激发;②价层中若干个能级相近的原子轨道“混杂”起来并重新组成一组新的轨道(称杂化轨道),这一过程称轨道的杂化。 (2)同一原子中能级相近的n个原子轨道,组合成n个杂化轨道,如sp杂化。 方法:两个轨道相加、相减 结果: 形状变化,一头大一头小,在大头方向上的分布比杂化前的s和p轨道大得多 方向相反
某原子成键时,在键合原子的作用下,原来原子的运动状态有可能发生变化,主要有两个方面①电子激发;②价层中若干个能级相近的原子轨道 混杂 起来并重新组成一组新的轨道(称杂化轨道),这一过程称轨道的杂化。 (2)同一原子中能级相近的n个原子轨道,组合成n个杂化轨道,如sp杂化。 方法:两个轨道相加、相减. 结果: 形状变化,一头大一头小,在大头方向上的分布比杂化前的s和p轨道大得多. 方向相反.")
28
(3)杂化轨道比原来杂化的轨道成键能力增强。形成的化学键键能大,使生成的分子更稳定
更大程度的重叠

29
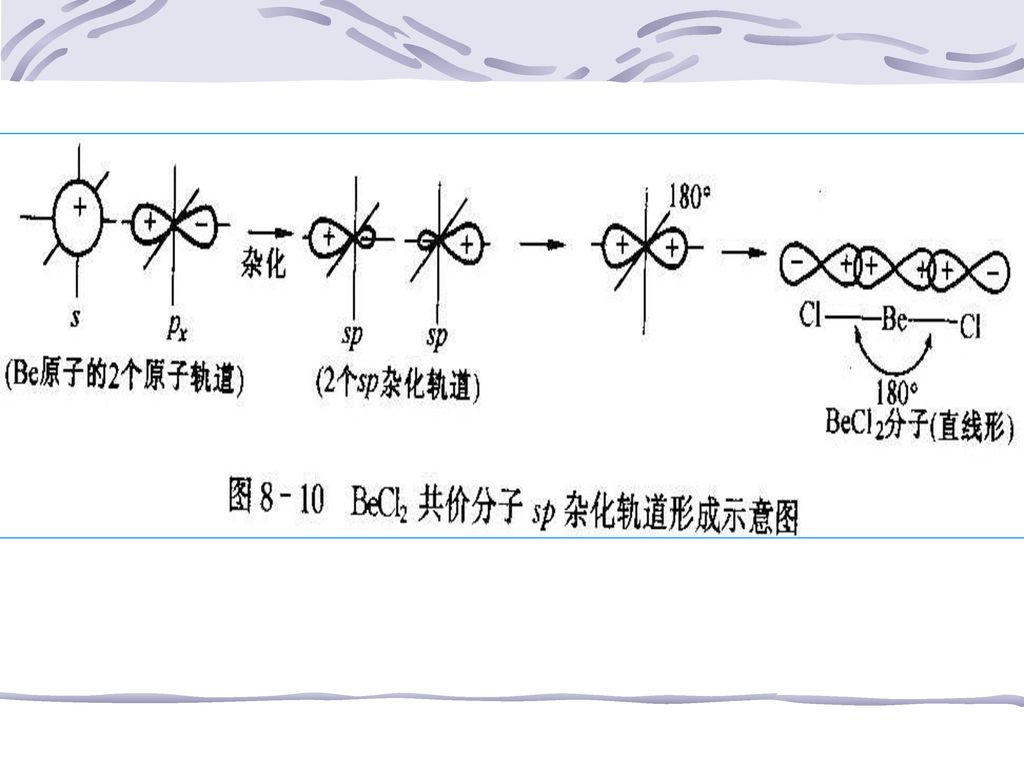
2、杂化类型与分子几何构型 (1)sp杂化:同一原子的1个ns+1个np杂化,得到两个sp杂化轨道 BeCl2 直线型共价分子,键角180°,键长、键能相等 杂化轨道解释: ①中心Be原子价层构型2s2,成键时,Be原子在键合Cl原子作用下,运动状态改变

30
周期表中ⅡB的某些共价化合物,如HgCl2也采取sp杂化。
<i>电子激发2s22p0→2s12p1,2s轨道与一个刚跃进电子的2p轨道发生sp杂化,形成两个等价sp轨道。每一个sp杂化轨道都含有1/2s+1/2p <ii>成键时,两个Cl原子分别以具有未成对电子的3p轨道与Be原子的2个sp轨道进行轨道重叠,电子配对,形成两个(p-sp)σ键,由于sp轨道s、p成份相同,所以与p轨道重叠后,重叠部分应相同,故BeCl2中两个键键能相同,键长相等 <iii>根据理论计算,这两个杂化轨道正好成180°夹角,即在同一直线上为最大限度重叠,Cl原子只能从直线两端进行重叠,Cl只能位于Be为中心的直线两端,故BeCl2为直线型分子 周期表中ⅡB的某些共价化合物,如HgCl2也采取sp杂化。
σ键,由于sp轨道s、p成份相同,所以与p轨道重叠后,重叠部分应相同,故BeCl2中两个键键能相同,键长相等. <iii>根据理论计算,这两个杂化轨道正好成180°夹角,即在同一直线上为最大限度重叠,Cl原子只能从直线两端进行重叠,Cl只能位于Be为中心的直线两端,故BeCl2为直线型分子. 周期表中ⅡB的某些共价化合物,如HgCl2也采取sp杂化。")
32
(2)sp2杂化:同一原子内1个ns+2个np 气态BF3: 三角形结构,3个B-F键等同,键角120° 每个sp2杂化轨道:1/3s+2/3p
sp2杂化:同一原子内1个ns+2个np 气态BF3: 三角形结构,3个B-F键等同,键角120° 每个sp2杂化轨道:1/3s+2/3p")
33
Fig.10-8 The planar configuration and hybrid orbital orientation of BF3 molecules

34
(3)SP3杂化:同一原子由一个s和3个p轨道混杂。每个sp3杂化轨道:1/4s+3/4p
CH4分子,正四面体构型,键角109°28′,四个C-H键等同.如CH4 ,SiH4 ,SiCl4 ,CCl4等

35
分子的几何构型 (4)不等性杂化 NH3分子, 键角107°18′ H2O分子, 键角104°45′
比较: sp2 120° sp3 109°28′ N、O均取SP3杂化,如N原子

36
键角减小原因 有一对电子未成键,因更靠近N原子,其电子云在N原子外占据较大空间,对三个N-H键的电子云有较大排斥作用,使键角被压缩到107°18′,故NH3为三角锥型。H2O为两对孤电子对,对成键电子对的排斥,键角被压得更小,故为104°45′。 不等性杂化: 由于孤电子对的电子云比较集中于中心原子附近,有更多的s轨道成分,所以四个sp3杂化轨道不完全等同。

37
d轨道参与的杂化: 第三周期及其后的元素原子。
杂化轨道的不足:预测分子几何构型未必能得到满意的结果. 新的理论,如价层电子对互斥理论(VSEPR)、分子轨道理论等。
、分子轨道理论等。")
38
6.4 分子轨道理论 液态和固态O2的顺磁性 光谱研究证明有H2+存在,
6.4 分子轨道理论 液态和固态O2的顺磁性 光谱研究证明有H2+存在, 分子轨道理论:说明很多分子的结构和反应性能问题上很成功,在共价键理论中,占有非常重要的位置。 理论假设出发点:成键电子是在整个分子区域内运动

39
(一)分子轨道理论M.O.的基本思路 把原子电子层结构的主要思路推广到分子体系而形成的一个分子结构理论,由原子的电子层结构引入分子中
(1)分子中的核心是分子中各原子核的平衡位 置构成的骨架 (2)分子核心之外,有若干个能级不相同的轨道,由低到高排列,越靠近分子核心,分子轨道的能量越低 (3)分子中的所有电子按两个原理一条规则填入分子轨道中。(分子中有能量相等的等价分子轨道时,分子尽可能占据不同的等价轨道,且自旋方向平行) (4)电子进入分子轨道后,若体系总能量降低,即能成键
分子中的核心是分子中各原子核的平衡位 置构成的骨架. (2)分子核心之外,有若干个能级不相同的轨道,由低到高排列,越靠近分子核心,分子轨道的能量越低. (3)分子中的所有电子按两个原理一条规则填入分子轨道中。(分子中有能量相等的等价分子轨道时,分子尽可能占据不同的等价轨道,且自旋方向平行) (4)电子进入分子轨道后,若体系总能量降低,即能成键.")
40
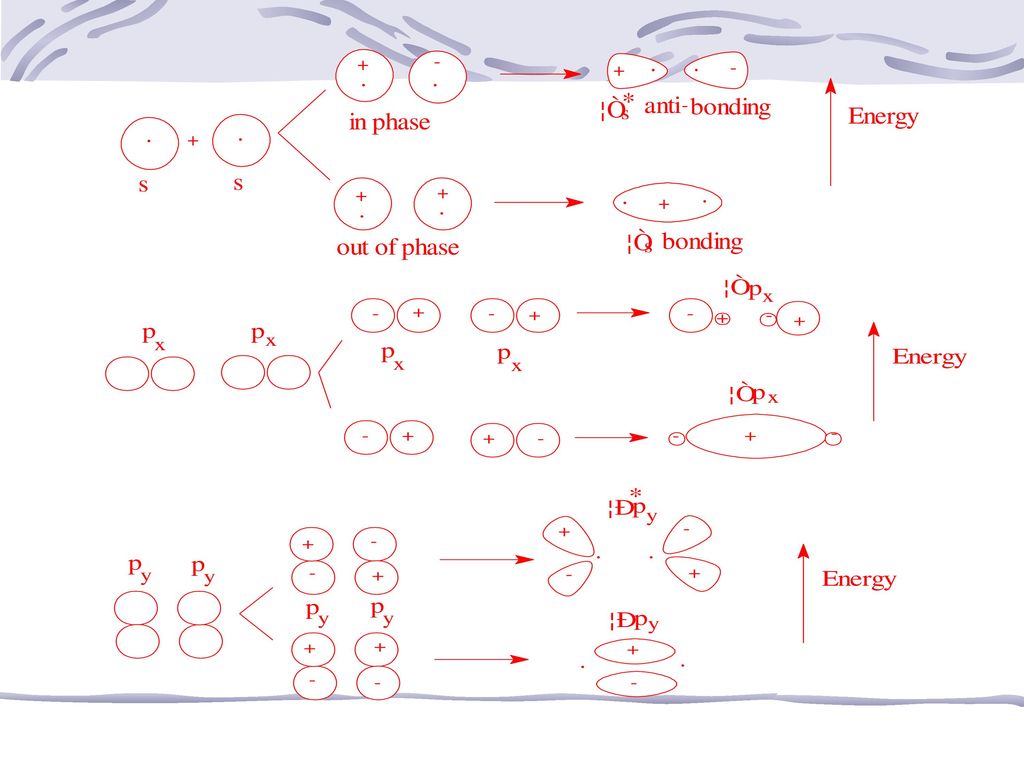
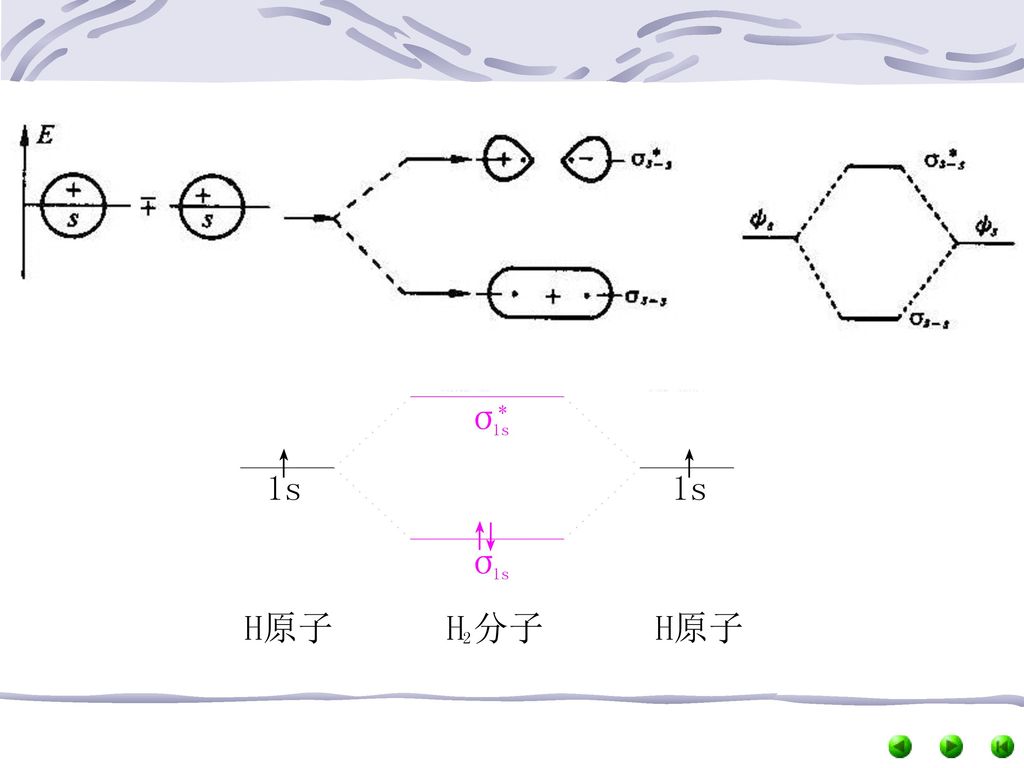
(二)分子轨道的形成 1.量子力学认为,M.O.是由A.O.组合而成 2.M.O.轨道数=参与组合的原子轨道总数 如当2个H原子组成H2分子时,两个H原子的1s轨道可组合成2个分子轨道 3.分子轨道的形状可通过A.O.的重叠,分别近似描述

42
1、s-s原子轨道的组合 反键分子轨道: 电子若原子核间有节面、能量较高分子轨道,对形成分子不利
成键分子轨道: 若进入原子核间电子云密度大、能量较低分子轨道,会使分子中原子键合 σ分子轨道: 分子轨道,其电子云沿键轴(两原子核连线)圆柱形对称分布。 σ﹡ns;σns σ轨道上的电子称σ电子 通过理论计算和实验测定可知,Eσ﹡ns>E ns>Eσns。 如H2分子轨道与H原子轨道能量关系图。 H2的分子轨道式:H2[(σ1s)2]
圆柱形对称分布。 σ﹡ns;σns σ轨道上的电子称σ电子. 通过理论计算和实验测定可知,Eσ﹡ns>E ns>Eσns。 如H2分子轨道与H原子轨道能量关系图。 H2的分子轨道式:H2[(σ1s)2]")
44
2、p-p原子轨道的组合 沿键轴方向的头碰头方式 肩并肩方式 σnpx成键分子轨道和σ﹡npx反键分子轨道
npz成键分子轨道和﹡npz反键分子轨道

45
2、P-P原子轨道的组合 可有“肩并肩”、“头碰头”两种方式组合 σ分子轨道 π分子轨道 (三)分子轨道的能级 每种分子的每个分子轨道都有确定的能量,不同分子的M.O.能量不同,可计算可测定P178图((a)(b))第一、二周期元素所组成的同核双原子分子能级图。 下面应用分子轨道理论来描述某些同核双原子分子的结构。

46
3 分子轨道的能级 第一、二周期除O2、F2以外的同核双原子分子的分子轨道能级
3 分子轨道的能级 第一、二周期除O2、F2以外的同核双原子分子的分子轨道能级 1s < *1s < 2s <*2s <2py = 2pz < 2px < *2py = *2pz < *2px O2、F2 1s < *1s < 2s <*2s <2px < 2py = 2pz < *2py = *2pz < *2px

47
例1:F2分子的结构根据6.24(a)图能级填电子:
分子轨道式: F2[KK(σ2s)2(σ*2s)2(σ2px)2(π2pg)2(π2pz)2(π*2py)2(π*2pz)2] F2形成的主要键,即(σ2p)2 即FσF键。 例2:N2分子结构 N2[KK(σ2s)2(σ*2s)2(π2pg)2(π2pz)2(σ2px)2] 故有三个键。 价键结构式和分子结构式
2(σ*2s)2(σ2px)2(π2pg)2(π2pz)2(π*2py)2(π*2pz)2] F2形成的主要键,即(σ2p)2 即FσF键。 例2:N2分子结构. N2[KK(σ2s)2(σ*2s)2(π2pg)2(π2pz)2(σ2px)2] 故有三个键。 价键结构式和分子结构式.")
48
4 分子轨道理论的应用: 1、推测分子的存在和阐明分子的结构 (1)H2+分子离子与Li2分子。 H2+[(σls)1]能存在,[H‧H]+中的键称单电子σ键。 Li2[KK(σ2s)2]体系能量降低,可能存在(气体中已观察到)[Li:Li] 称单σ键。 (2)Be2分子和Ne2分子 Be2[KK(σ2s)2(σ*2s)2] Ne2[KK(σ2s)2(σ*2s)2(σ2px)2(π2py)2(π2pz)2(π*2py)2(π*2pz)2(σ*2px)2]能量抵消 ∴Be2、Ne2不是很不稳定就是根本不存在。事实上,到目前未发现它们。
![4 分子轨道理论的应用: 1、推测分子的存在和阐明分子的结构. (1)H2+分子离子与Li2分子。 H2+[(σls)1]能存在,[H‧H]+中的键称单电子σ键。 Li2[KK(σ2s)2]体系能量降低,可能存在(气体中已观察到)[Li:Li] 称单σ键。](http://slidesplayer.com/slide/11388548/61/images/48/4+%E5%88%86%E5%AD%90%E8%BD%A8%E9%81%93%E7%90%86%E8%AE%BA%E7%9A%84%E5%BA%94%E7%94%A8%EF%BC%9A+1%E3%80%81%E6%8E%A8%E6%B5%8B%E5%88%86%E5%AD%90%E7%9A%84%E5%AD%98%E5%9C%A8%E5%92%8C%E9%98%90%E6%98%8E%E5%88%86%E5%AD%90%E7%9A%84%E7%BB%93%E6%9E%84.+%EF%BC%881%EF%BC%89H2%2B%E5%88%86%E5%AD%90%E7%A6%BB%E5%AD%90%E4%B8%8ELi2%E5%88%86%E5%AD%90%E3%80%82+H2%2B%5B%28%CF%83ls%291%5D%E8%83%BD%E5%AD%98%E5%9C%A8%EF%BC%8C%5BH%E2%80%A7H%5D%2B%E4%B8%AD%E7%9A%84%E9%94%AE%E7%A7%B0%E5%8D%95%E7%94%B5%E5%AD%90%CF%83%E9%94%AE%E3%80%82+Li2%5BKK%28%CF%832s%292%5D%E4%BD%93%E7%B3%BB%E8%83%BD%E9%87%8F%E9%99%8D%E4%BD%8E%EF%BC%8C%E5%8F%AF%E8%83%BD%E5%AD%98%E5%9C%A8%EF%BC%88%E6%B0%94%E4%BD%93%E4%B8%AD%E5%B7%B2%E8%A7%82%E5%AF%9F%E5%88%B0%EF%BC%89%5BLi%3ALi%5D+%E7%A7%B0%E5%8D%95%CF%83%E9%94%AE%E3%80%82.jpg "(2)Be2分子和Ne2分子. Be2[KK(σ2s)2(σ*2s)2] Ne2[KK(σ2s)2(σ*2s)2(σ2px)2(π2py)2(π2pz)2(π*2py)2(π*2pz)2(σ*2px)2]能量抵消 ∴Be2、Ne2不是很不稳定就是根本不存在。事实上,到目前未发现它们。")
49
(3)He2分子和He2+分子离子。 He2[(σls)2(σ*ls)2],所以He2不存在,因此稀有气体都是单原子分子。 He2+[(σls)2(σ*ls)1],体系总能量下降,可以存在。为了区别于单电子σ键,把[He He]+分子离子中的化学键称三电子σ键。 2、描述分子的结构稳定性 键级= 一般键级越大,键能越大,分子越稳定。但只是定性推断,有例外。
![(3)He2分子和He2+分子离子。 He2[(σls)2(σ*ls)2],所以He2不存在,因此稀有气体都是单原子分子。 He2+[(σls)2(σ*ls)1],体系总能量下降,可以存在。为了区别于单电子σ键,把[He He]+分子离子中的化学键称三电子σ键。](http://slidesplayer.com/slide/11388548/61/images/49/%EF%BC%883%EF%BC%89He2%E5%88%86%E5%AD%90%E5%92%8CHe2%2B%E5%88%86%E5%AD%90%E7%A6%BB%E5%AD%90%E3%80%82+He2%5B%28%CF%83ls%292%28%CF%83%2Als%292%5D%EF%BC%8C%E6%89%80%E4%BB%A5He2%E4%B8%8D%E5%AD%98%E5%9C%A8%EF%BC%8C%E5%9B%A0%E6%AD%A4%E7%A8%80%E6%9C%89%E6%B0%94%E4%BD%93%E9%83%BD%E6%98%AF%E5%8D%95%E5%8E%9F%E5%AD%90%E5%88%86%E5%AD%90%E3%80%82+He2%2B%5B%28%CF%83ls%292%28%CF%83%2Als%291%5D%EF%BC%8C%E4%BD%93%E7%B3%BB%E6%80%BB%E8%83%BD%E9%87%8F%E4%B8%8B%E9%99%8D%EF%BC%8C%E5%8F%AF%E4%BB%A5%E5%AD%98%E5%9C%A8%E3%80%82%E4%B8%BA%E4%BA%86%E5%8C%BA%E5%88%AB%E4%BA%8E%E5%8D%95%E7%94%B5%E5%AD%90%CF%83%E9%94%AE%EF%BC%8C%E6%8A%8A%5BHe+He%5D%2B%E5%88%86%E5%AD%90%E7%A6%BB%E5%AD%90%E4%B8%AD%E7%9A%84%E5%8C%96%E5%AD%A6%E9%94%AE%E7%A7%B0%E4%B8%89%E7%94%B5%E5%AD%90%CF%83%E9%94%AE%E3%80%82.jpg "2、描述分子的结构稳定性. 键级= 一般键级越大,键能越大,分子越稳定。但只是定性推断,有例外。")
50
3、预言分子的顺磁性与反磁性 物质磁性实验发现,凡有未成对电子的分子,在外加磁场中必须磁场方向排列,分子的这种性质叫顺磁性,具有这种性质的物质称顺磁性物质,反之,为反磁性。 O2[KK(σ2s)2(σ*2s)2(σ2px)2(π2py)2(π2pz)2(π*2py)2(π*2pz)1] 所以O2分子具有顺磁性。
![3、预言分子的顺磁性与反磁性 物质磁性实验发现,凡有未成对电子的分子,在外加磁场中必须磁场方向排列,分子的这种性质叫顺磁性,具有这种性质的物质称顺磁性物质,反之,为反磁性。 O2[KK(σ2s)2(σ*2s)2(σ2px)2(π2py)2(π2pz)2(π*2py)2(π*2pz)1]](http://slidesplayer.com/slide/11388548/61/images/50/3%E3%80%81%E9%A2%84%E8%A8%80%E5%88%86%E5%AD%90%E7%9A%84%E9%A1%BA%E7%A3%81%E6%80%A7%E4%B8%8E%E5%8F%8D%E7%A3%81%E6%80%A7+%E7%89%A9%E8%B4%A8%E7%A3%81%E6%80%A7%E5%AE%9E%E9%AA%8C%E5%8F%91%E7%8E%B0%EF%BC%8C%E5%87%A1%E6%9C%89%E6%9C%AA%E6%88%90%E5%AF%B9%E7%94%B5%E5%AD%90%E7%9A%84%E5%88%86%E5%AD%90%EF%BC%8C%E5%9C%A8%E5%A4%96%E5%8A%A0%E7%A3%81%E5%9C%BA%E4%B8%AD%E5%BF%85%E9%A1%BB%E7%A3%81%E5%9C%BA%E6%96%B9%E5%90%91%E6%8E%92%E5%88%97%EF%BC%8C%E5%88%86%E5%AD%90%E7%9A%84%E8%BF%99%E7%A7%8D%E6%80%A7%E8%B4%A8%E5%8F%AB%E9%A1%BA%E7%A3%81%E6%80%A7%EF%BC%8C%E5%85%B7%E6%9C%89%E8%BF%99%E7%A7%8D%E6%80%A7%E8%B4%A8%E7%9A%84%E7%89%A9%E8%B4%A8%E7%A7%B0%E9%A1%BA%E7%A3%81%E6%80%A7%E7%89%A9%E8%B4%A8%EF%BC%8C%E5%8F%8D%E4%B9%8B%EF%BC%8C%E4%B8%BA%E5%8F%8D%E7%A3%81%E6%80%A7%E3%80%82+O2%5BKK%28%CF%832s%292%28%CF%83%2A2s%292%28%CF%832px%292%28%CF%802py%292%28%CF%802pz%292%28%CF%80%2A2py%292%28%CF%80%2A2pz%291%5D.jpg "所以O2分子具有顺磁性。")
51
优缺点: 价键理论:(1)简明直观;(2)价键概念突出;(3)在描述分子的几何构型方面有独到之处,容易掌握。但认为是定域键,且只有配对才能成键,使它的应用范围变窄,对许多分子的结构和性能不能给予确切解释。 M.O.理论,把电子分布统筹安排,使分子具有整体性(非定域键);把成键条件放宽,有单电子键,故应用范围变宽,但它的价键概念不明确,计算方法复杂,一般人不易运用和掌握;在描述分子几何构型方面也不够直观。
;把成键条件放宽,有单电子键,故应用范围变宽,但它的价键概念不明确,计算方法复杂,一般人不易运用和掌握;在描述分子几何构型方面也不够直观。")
52
6.5 分子间力和氢键 6.5.1 分子的极性和变形性 1、分子的极性
每个分子中正、负电荷总量相等,整个分子是电中性的。但对每一种电荷量来说,都可设想一个集中点,称“电荷中心”。分子中若正、负电荷中心不重合在同一点上,那么这两个中心又可称作分子的两个极(正极和负极),这样的分子就具有极性
,这样的分子就具有极性.")
53
(1)同核双原子分子 非极性键,正、负电荷重合,非极性分子 (2)不同核双原子分子 极性键,正、负电荷不重合,极性分子 (3)多原子分子 分子组成和分子的几何构型 H2O,CO2,CH4,NH3 键的极性—电子对是否偏移(电负性) 分子的极性—整个分子正、负电荷中心是否重合
 分子的极性—整个分子正、负电荷中心是否重合.")
54
按极性强弱分类 离子型分子,极性分子,非极性分子 分子偶极矩(μ):衡量分子极性的大小。定义为 正或负电荷中心上的电荷量q与正负电荷中心的 间距d。单位:库·米(c·m) μ=q.d d又称偶极长度 应用 (1)μ=O非极性分子;μ≠0极性分子。 (2)μ越大,极性越强。比较分子极性的强弱 (3)用μ验证或判断某些分子的几何构型。如 CS2的μ=0,直线型分子
μ=O非极性分子;μ≠0极性分子。 (2)μ越大,极性越强。比较分子极性的强弱. (3)用μ验证或判断某些分子的几何构型。如 CS2的μ=0,直线型分子.")
55
2、分子变形性 分子的变形极化:在外加电场作用下,分子正、负电荷中心发生相对位移,分子产生形变。此时,由于正负电荷彼此分离产生的偶极,为诱导偶极μ诱导 分子的变形性:分子中因电子云和核发生相对位移而使分子外形发生变化的性质 μ诱导=α×E α称分子的诱导极化率(极化率) 外电场越强,分子变形越显著,诱导极化率越大,μ越大,电场撤离,分子变为非极性
 外电场越强,分子变形越显著,诱导极化率越大,μ越大,电场撤离,分子变为非极性.")
56
固有偶极(永久偶极): 极性分子本身存在的偶极
在外电场作用下,极性分子会取一定方向排列,这一过程称为分子的定向极化。同时产生诱导偶极,分子极性增强。 极性分子的偶极=固有偶极+诱导偶极 极性分子在电场中的极化包括分子的定向极化和变形极化两方面。

57
6.5.2 分子间力 1、分子间的相互吸引作用 (1)非极性分子间
分子不断地运动→电子云和原子核之间瞬时相对位移→正负电荷中心暂时不重合→瞬时偶极 色散力——分子间由瞬时偶极而产生的作用力,非极性分子间正是因此而形成固体或液体

58
(2)非极性分子和极性分子间 诱导力:非极性分子在极性分子固有偶极作用下会发生变形极化,产生诱导偶极,这种由诱导偶极与固有偶极间产生的作用力 极性分子也会出现瞬时偶极,这样在非极性分子与极性分子间还会有色散力。 诱导力+色散力

59
(3)极性分子间 取向力:极性分子互相接近时,因固有偶极而发生定向极化,由于固有偶极的取向而产生的作用力 极性分子定向排列后还会进一步产生变形极化,产生诱导偶极,因此还有诱导力,也有色散力 取向力+诱导力+色散力

60
2、分子间力的特点和影响因素 特点(五类) (1)本质是一种电性作用力(静电吸引) (2)作用范围:是短程力,作用范围仅几百皮米,当分子间距离为分子本身直径的4~5倍时,作用力迅速减弱 (3)作用能:一般只有几-几十kJ/mol,比化学键键能小1-2个数量级,但对由共价型分子组成的物质的一些物理性质影响很大 (4)一般没有饱和性和方向性 (5)对大多数分子,色散力是主要的,一般相对大小,色散力>>取向力>诱导力
作用能:一般只有几-几十kJ/mol,比化学键键能小1-2个数量级,但对由共价型分子组成的物质的一些物理性质影响很大. (4)一般没有饱和性和方向性. (5)对大多数分子,色散力是主要的,一般相对大小,色散力>>取向力>诱导力.")
61
影响因素: ①分子间距离(主要因素),距离增大,作用力迅速减弱 ②取向力还与温度和分子的极性强弱有关:温度升高,取向难,取向力减弱,分子偶极矩越大,取向力越强 ③诱导力还与极性分子极性强弱和非极性分子的变形性大小有关:极性分子偶极矩越大,非极性分子极化率越大,诱导力越强 ④色散力主要与分子的变形性有关,即分子极化率越大,色散力越强

62
3、分子间力对物质物理性质的影响: 主要影响熔、沸点、气化热、熔化热。 液态物质分子间力越大,气化热越大,沸点越高 固态物质分子间力越大,熔化热越大,熔点越高 一般说:结构相似的同系列物质(如X2)相对分子质量越大,分子变形性也越大,分子间力越强,物质的熔、沸点就越高
相对分子质量越大,分子变形性也越大,分子间力越强,物质的熔、沸点就越高.")
63
相对分子质量相等(如H3PO4、H2SO4)或近似而体积大的有较大变形性熔、沸点相对较高。
溶质或溶剂(若它们是同系列,如有机物中)的极化率α越大,分子变形性和分子间的力越大,溶解度越大。 分子极性小的(如聚乙烯、聚异丁烯等)分子间力小,硬度不大,含有极性基因的有机玻璃等物,分子间力较大,具有一定的硬度。
的极化率α越大,分子变形性和分子间的力越大,溶解度越大。 分子极性小的(如聚乙烯、聚异丁烯等)分子间力小,硬度不大,含有极性基因的有机玻璃等物,分子间力较大,具有一定的硬度。")
64
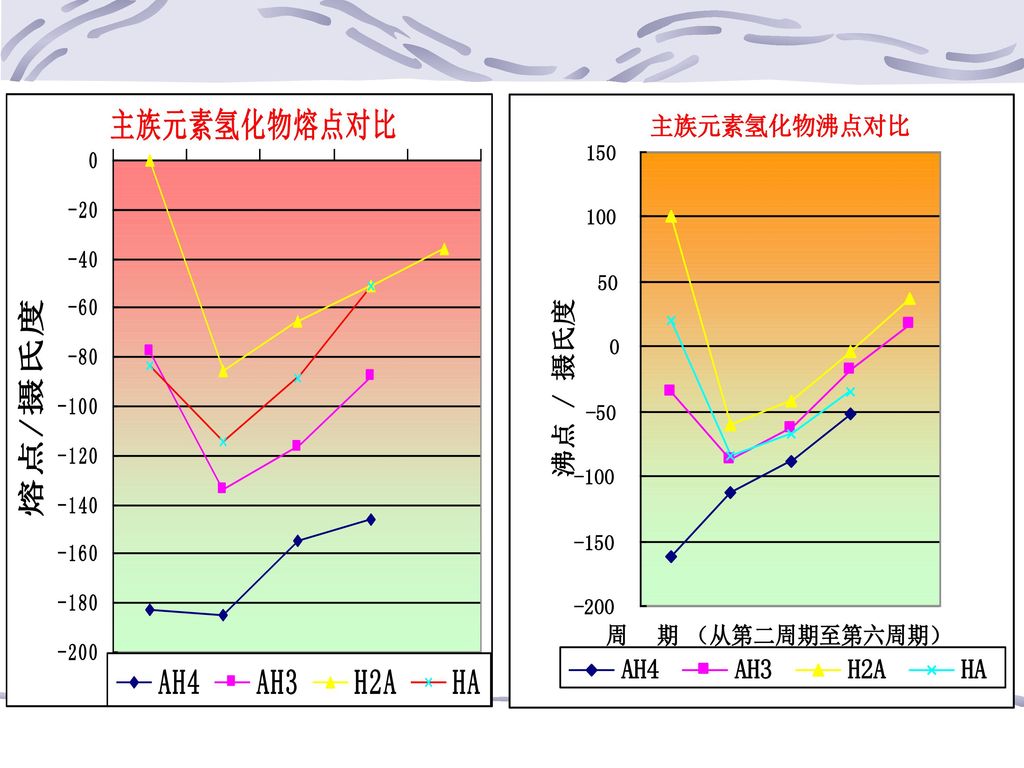
(三)氢键 在HX熔、沸点变化出现反常,这是因除分子间力外,还有氢键 1、氢键的形成 如HF分子中,半径减小,它内层电子的带部分正电荷的氢原子与附近的HF分子中含孤电子对并带部分负电荷的F原子间产生的静电吸引力,即氢键 同种分子间可产生氢键,不同种分子间也可产生氢键。如NH3和H2O 氢键通式:X—H……Y:X、Y表示F/O/N等电负性大而原子半径小的非金属原子

65
冰的晶体结构:(小球代表氢原子,大球代表氧原子,实线代表H—O键,虚线代表氢键)
")
66
2、氢键的强度 3、分子内氢键 如HNO3,如硝基苯酚等
可用键能来衡量,氢键功能—拆开单位物质的量的H…Y键所需的能量。一般 42kJ·mol-1以下,比共价键键能小很多,而与分子间力更接近,例如H2O中, =463 kJ·mol-1,而氢键键能仅为18.83 kJ·mol-1 3、分子内氢键 如HNO3,如硝基苯酚等

67
4、氢键形成对物质性质的影响 液、晶态或气态物质中,如HF气、液、固态均有 能形成氢键的物质很多,如水合物、氨合物、无机酸和某些有机化合物,如DNA,双螺旋体间存在氢键。 (1)熔、沸点 分子间有氢键存在的物质比同系列中无氢键物质熔、沸点高,如HX。 分子内氢键,熔、沸点常降低,如有分子内氢键的邻硝基酚熔点(45℃)比有分子间氢键的间位硝基苯酚(以熔点定96℃)和对位硝基苯酚的熔点(114℃)都低
比有分子间氢键的间位硝基苯酚(以熔点定96℃)和对位硝基苯酚的熔点(114℃)都低.")
69
(2)溶解度:在极性溶剂中,如果溶液和溶剂分子间形成氢键,溶质的溶解度增大,如HF和NH3在水中溶解度较大。
(3)粘度:分子间有氢键的液体,一般粘度较大,如甘油、H3PO4、浓硫酸等多羟基化合物。
粘度:分子间有氢键的液体,一般粘度较大,如甘油、H3PO4、浓硫酸等多羟基化合物。")
70
(4)密度:液体分子间若形成氢键,有可能发生缔合现象,如液态HF中除简单HF分子外,还有通过氢键联系的复杂分子:nHF (HF)n
由若干个简单分子联成复杂分子而不会改变原物质化学性质的现象——分子的缔合,又如 nH2O (H2O) 降温有利于分子的缔合,降至0℃,全部水分子结合成巨大的缔合物——冰。
 降温有利于分子的缔合,降至0℃,全部水分子结合成巨大的缔合物——冰。")
71
§7-6 离子极化对物质性质的影响 影响离子性质的因素 离子电荷、离子半径、电子构型 复习离子的电子构型 表 p211

72
1 离子极化:当离子置于电场中,离子的原子核受到正电场的排斥和负电场的吸引,离子中的电子受到正电场的吸引和负电场的排斥,发生形变而产生诱导偶极。
影响离子极化的影响因素:离子的极化力和离子的变形性

73
2 影响离子的极化力的因素 离子的电荷越多、离子半径越小、产生的电场强度越大,极化能力越强 电子构型 (18、18+2、2)>(9~17)>(8)
>(9~17)>(8)")
74
3 影响离子的变形性的因素 根本: 原子核对外层电子的引力越小,变形性越大 离子半径大,变形性大 阴离子比相应电子构型的阳离子容易变形 正电荷越多,变形性越小(极化力大) 负电荷越多,变形性越大(极化力小) 电子构型:d电子数越多,变形性越大 (18、18+2、 9~17)> (2、8)
> (2、8)")
75
离子变形性大小由离子极化率量度 离子极化率=诱导偶极E 表7.4 p213

76
4 离子极化的规律 阴离子半径相同时,阳离子电荷越多,阴离子越容易被极化,产生的诱导偶极越大 阳离子电荷相同时,阳离子越大,对阴离子的极化力越小,产生的诱导偶极越小 阳离子的电荷相同、大小相近时,阴离子越大,越容易被极化,产生的诱导偶极越大

77
(三)离子极化对物质结构的性质的影响 离子极化对化学键型的影响
当极化力强、变形性大的阳离子与变形性大的阴离子相互作用时,导致阳、阴离子外层轨道发生重叠,键长缩短,键的极性减弱,化学键从离子键向共价键过渡。 AgF离子键,AgCl、AgBr过渡键型,AgI共价键

78
二. 极化使离子键向共价键过渡 解释下列现象: 1.为什么AlCl3的熔点小于NaCl,且能溶在有机溶剂中?
2.为什么卤化银的溶解度逐渐降低? 3.为什么铬酸钾是黄色而铬酸银为砖红色? 极化后,电子能级改变,基态和激发态的能级差变小,吸收可见光就可激发。


 如何区分元素和微粒(分子、原子、离子) 1 .概念的范畴不同 元素 —— 宏观概念, 只论种类不论个数 微粒 —— 微观概念, 既论种类又论个数 2 .决定种类的因素不同 元素 —— 由核电荷数 ( 即质子数 ) 决定 (如 16.>")
. 年度 90 -190-291-191-292-192-2 總題數 356565 章 1 234567 總題數 (90 ~ 92) 6 444552.>")
 晶体自范性的本质 : 是晶体中粒子微观空间里 呈现周期性的有序排列的宏观表象. (2) 晶体自范性的条件之一 : 生长速率适当. 生长速率适当. 自范性微观结构 晶体有 ( 能自发呈现多面体外 形 )>")









 提出离子键理论。 一 离子键的形成>")






