Download presentation
1
ultraviolet –visible spectrometry, UV-VIS
仪器分析 第四章 紫外可见分光光度法 ultraviolet –visible spectrometry, UV-VIS

2
本 章 内 容 仪器分析 §1 概述 §2 电磁辐射 §3 分子吸收光谱 §4 光的吸收定律 §5 分光光度计 §6 测量条件的选择
§7 定性及定量分析 §8 催化动力学分光光度法 §9 提高灵敏度和准确度 §10 分光光度法的应用 本 章 内 容

3
一、光分析法及其特点 optical analysis and its characteristics
§1 概述 一、光分析法及其特点 optical analysis and its characteristics 光分析法:基于电磁辐射能量与待测物质相互作用后所产生的辐射信号与物质组成及结构关系所建立起来的分析方法; 电磁辐射范围:射线~无线电波所有范围; 相互作用方式:吸收、发射、反射、折射、散射、干涉、衍射等; 光分析法在研究物质组成、结构表征、表面分析等方面具有其他方法不可区代的地位;

4
三个基本过程: 基本特点: (1)能源提供能量; (2)能量与被测物之间的相互作用; (3)产生信号检测。
(1)所有光分析法均包含三个基本过程; (2)选择性测量,不涉及混合物分离(不同于色谱分析); (3)涉及大量光学元器件。
所有光分析法均包含三个基本过程; (2)选择性测量,不涉及混合物分离(不同于色谱分析); (3)涉及大量光学元器件。")
5
二、光分析法分类 type of optical analysis
光谱法——基于物质与辐射能作用时,分子发生能级跃迁而产生的发射、吸收或散射的波长或强度进行分析的方法; 原子光谱、分子光谱 原子光谱(线性光谱):最常见的三种 基于原子外层电子跃迁的原子吸收光谱(AAS); 原子发射光谱(AES)、原子荧光光谱(AFS); 基于原子内层电子跃迁的 X射线荧光光谱(XFS); 基于原子核与射线作用的穆斯堡谱;
:最常见的三种. 基于原子外层电子跃迁的原子吸收光谱(AAS); 原子发射光谱(AES)、原子荧光光谱(AFS); 基于原子内层电子跃迁的 X射线荧光光谱(XFS); 基于原子核与射线作用的穆斯堡谱;")
6
分子光谱(带状光谱): 非光谱法: 基于分子中电子能级、振-转能级跃迁; 紫外光谱法(UV); 红外光谱法(IR);
分子荧光光谱法(MFS); 分子磷光光谱法(MPS); 核磁共振波谱(NMR); 非光谱法: 不涉及能级跃迁,物质与辐射作用时,仅改变传播方向等物理性质;偏振法、干涉法、旋光法等;
; 分子磷光光谱法(MPS); 核磁共振波谱(NMR); 非光谱法: 不涉及能级跃迁,物质与辐射作用时,仅改变传播方向等物理性质;偏振法、干涉法、旋光法等;")
7
光分析法 光谱分析法 非光谱分析法 折 射 法 圆 二 色 性 X 线 衍 干 涉 旋 光 原子光谱分析法 分子光谱分析法 原 子 吸 收
发 射 荧 X 线 紫 外 光 谱 法 红 分 子 荧 磷 核 磁 共 振 波

8
原 子 发 射 吸 收 荧 光 紫 外 可 见 红 分 子 荧 光 磷 核 磁 共 振 化 学 发 光谱分析法 吸收光谱法 发射光谱法
X 线 紫 外 可 见 红 分 子 荧 光 磷 核 磁 共 振 化 学 发 光谱分析法 吸收光谱法 发射光谱法 原子光谱法 分子光谱法 原 子 发 射 荧 光 分 磷 X 线 化 学 原 子 吸 收 紫 外 可 见 红 核 磁 共 振

9
三、各种光谱分析法简介 a brief introduction of optical analysis
1.原子发射光谱分析法 以火焰、电弧、等离子炬等作为光源,使气态原子的外层电子受激发射出特征光谱进行定量分析的方法。 2.原子吸收光谱分析法 利用特殊光源发射出待测元素的共振线,并将溶液中离子转变成气态原子后,测定气态原子对共振线吸收而进行的定量分析方法。

10
3.原子荧光分析法 气态原子吸收特征波长的辐射后,外层电子从基态或低能态跃迁到高能态,在10-8s后跃回基态或低能态时,发射出与吸收波长相同或不同的荧光辐射,在与光源成90度的方向上,测定荧光强度进行定量分析的方法。 4.分子荧光分析法 某些物质被紫外光照射激发后,在回到基态的过程中发射出比原激发波长更长的荧光,通过测量荧光强度进行定量分析的方法。

11
5. 分子磷光分析法 6. X射线荧光分析法 7. 化学发光分析法
处于第一最低单重激发态分子以无辐射弛豫方式进入第一三重激发态,再跃迁返回基态发出磷光。测定磷光强度进行定量分析的方法。 6. X射线荧光分析法 原子受高能辐射,其内层电子发生能级跃迁,发射出特征X射线( X射线荧光),测定其强度可进行定量分析。 7. 化学发光分析法 利用化学反应提供能量,使待测分子被激发,返回基态时发出一定波长的光,依据其强度与待测物浓度之间的线性关系进行定量分析的方法。
,测定其强度可进行定量分析。 7. 化学发光分析法. 利用化学反应提供能量,使待测分子被激发,返回基态时发出一定波长的光,依据其强度与待测物浓度之间的线性关系进行定量分析的方法。")
12
8. 紫外吸收光谱分析法 9.红外吸收光谱分析法 10.核磁共振波谱分析法
利用溶液中分子吸收紫外和可见光产生跃迁所记录的吸收光谱图,可进行化合物结构分析,根据最大吸收波长强度变化可进行定量分析。 9.红外吸收光谱分析法 利用分子中基团吸收红外光产生的振动-转动吸收光谱进行定量和有机化合物结构分析的方法。 10.核磁共振波谱分析法 在外磁场的作用下,核自旋磁矩与磁场相互作用而裂分为能量不同的核磁能级,吸收射频辐射后产生能级跃迁,根据吸收光谱可进行有机化合物结构分析 。

13
11.顺磁共振波谱分析法 12.旋光法 13.衍射法 X射线衍射:研究晶体结构,不同晶体具有不同衍射图。
在外磁场的作用下,电子的自旋磁矩与磁场相互作用而裂分为磁量子数不同的磁能级,吸收微波辐射后产生能级跃迁,根据吸收光谱可进行结构分析 。 12.旋光法 溶液的旋光性与分子的非对称结构有密切关系,可利用旋光法研究某些天然产物及配合物的立体化学问题,旋光计测定糖的含量。 13.衍射法 X射线衍射:研究晶体结构,不同晶体具有不同衍射图。 电子衍射:电子衍射是透射电子显微镜的基础,研究物质的内部组织结构。

14
四、光分析方法的进展 development of optical analysis
1. 采用新光源,提高灵敏度 级联光源:电感耦合等离子体-辉光放电;激光蒸发-微波等离子体 2. 联用技术 电感耦合高频等离子体(ICP)—质谱 激光质谱:灵敏度达10-20 g 3. 新材料 光导纤维传导,损耗少;抗干扰能力强;
—质谱. 激光质谱:灵敏度达10-20 g. 3. 新材料. 光导纤维传导,损耗少;抗干扰能力强;")
15
4. 交叉 5. 检测器的发展 光二极激光器代替空心阴极灯,使原子吸收可进行多元素同时测定; 电致发光分析;光导纤维电化学传感器
电荷耦合阵列检测器光谱范围宽、量子效率高、线性范围宽、多道同时数据采集、三维谱图,将取代光电倍增管; 光二极激光器代替空心阴极灯,使原子吸收可进行多元素同时测定;

16
三种光分析法测量过程示意图

17
分光光度法:基于物质对光的选择性吸收的分析方法
类型:比色法 可见分光光度法(visible spectrophotometry) 紫外分光光度法(ultraviolet spectrophotometry)
 紫外分光光度法(ultraviolet spectrophotometry)")
18
特点: (1) 具有较高的灵敏度,通常所测试液的浓度下限达10-5~10-6 mol·L-1,适用于微量组分的测定。
(2) 分光光度法测定的相对误差约为2%~5%。 (3) 测定迅速,仪器操作简单,价格便宜,应用广泛 (4) 几乎所有的无机物质和许多有机物质的微量成分都能用此法进行测定。 (5) 常用于化学平衡等的研究。
 分光光度法测定的相对误差约为2%~5%。 (3) 测定迅速,仪器操作简单,价格便宜,应用广泛. (4) 几乎所有的无机物质和许多有机物质的微量成分都能用此法进行测定。 (5) 常用于化学平衡等的研究。")
19
光子运动具有微观粒子的波动性和粒子性,即波粒二象性。 光子的能量与频率或波长的关系:
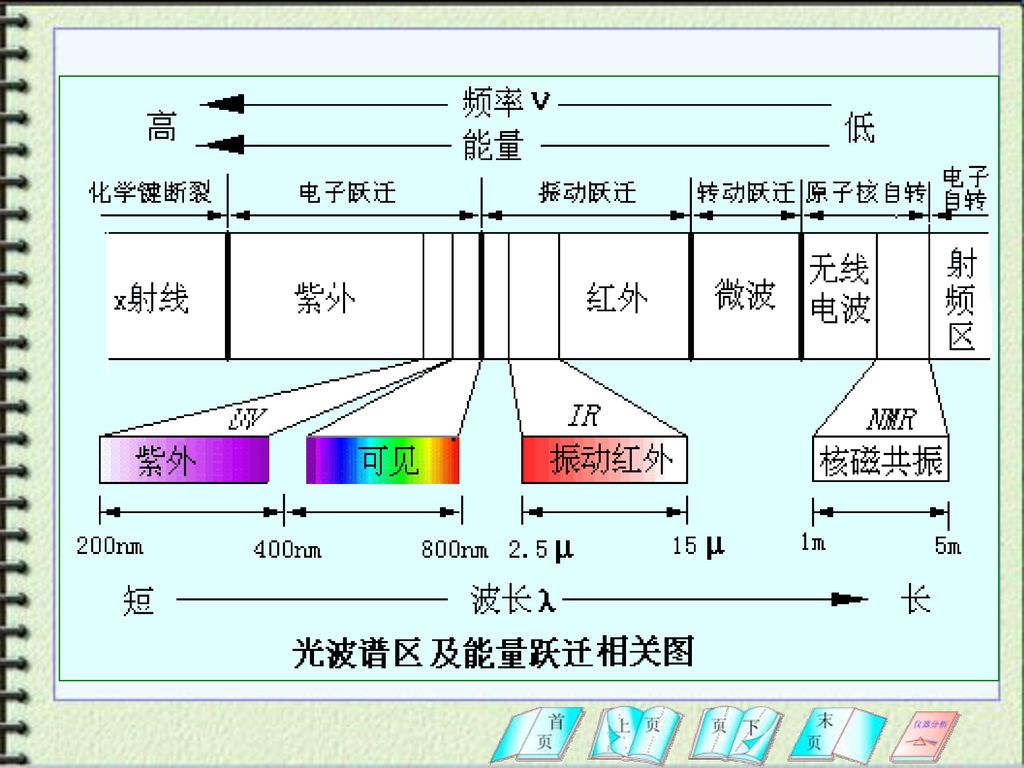
§2 电磁辐射 光是一种电磁辐射(electromagnetic radiation),又称电磁波,是一种以巨大速度通过空间传播的光量子流,它的基本单位是光子。 光子运动具有微观粒子的波动性和粒子性,即波粒二象性。 光子的能量与频率或波长的关系: 电磁辐射按波长顺序排列称为电磁波谱(electromagnetic spectrum)。电磁波谱包括的波长范围很宽 。
,又称电磁波,是一种以巨大速度通过空间传播的光量子流,它的基本单位是光子。 光子运动具有微观粒子的波动性和粒子性,即波粒二象性。 光子的能量与频率或波长的关系: 电磁辐射按波长顺序排列称为电磁波谱(electromagnetic spectrum)。电磁波谱包括的波长范围很宽 。")
21
电磁波谱 辐射类型 波长范围 光谱类型 跃迁类型 γ射线 0.005~0.14nm γ射线光谱 核能级 X射线 0.01~10nm
内层电子能级 远紫外光 近紫外光 可见光 近红外光 中红外光 远红外光 微波 无线电波 10~200nm 200~400nm 400~760nm 0.76~2.5μm 2.5~50μm 50~1000μm 0.1~100cm 1~1000m 真空紫外光谱 紫外光谱 可见光谱 红外光谱 微波波谱 核磁共振谱 原子及分子的价电子或成键电子能级 分子振动能级 分子振动能级和转动能级 分子转动能级 分子转动能级和电子自旋能级 磁场诱导核自旋能级

22
电磁辐射与物质的相互作用 (1) 吸收 物质选择性吸收特定频率的辐射能,并从低能级跃迁到高能级;
(1) 吸收 物质选择性吸收特定频率的辐射能,并从低能级跃迁到高能级; (2) 发射 将吸收的能量以光的形式释放出; (3) 散射 丁铎尔散射和分子散射(瑞利散射和拉曼散射) (4) 折射 折射是光在两种介质中的传播速度不同; (5) 反射 (6) 干涉 干涉现象; (7) 衍射 光绕过物体而弯曲地向他后面传播的现象; (8) 偏振 只在一个固定方向有振动的光称为平面偏振光
 吸收 物质选择性吸收特定频率的辐射能,并从低能级跃迁到高能级; (2) 发射 将吸收的能量以光的形式释放出; (3) 散射 丁铎尔散射和分子散射(瑞利散射和拉曼散射) (4) 折射 折射是光在两种介质中的传播速度不同; (5) 反射. (6) 干涉 干涉现象; (7) 衍射 光绕过物体而弯曲地向他后面传播的现象; (8) 偏振 只在一个固定方向有振动的光称为平面偏振光.")
23
§3 分子吸收光谱 一、分子吸收光谱的产生 M + 热 M + h M * M + 荧光或磷光 基态 激发态 E0 (△E) E1
基态 激发态 E (△E) E1 E = E E0 = h 选择性吸收;量子化 ; 分子结构的复杂性使其对不同波长光的吸收程度不同; 光的互补:蓝 黄
 E1. E = E1 - E0 = h 选择性吸收;量子化 ; 分子结构的复杂性使其对不同波长光的吸收程度不同; 光的互补:蓝 黄.")
24
物质颜色与吸收光颜色的互补关系

25
1. 过程:运动的分子外层电子--------吸收外来辐射------电子能级跃迁-----形成分子吸收光谱。 2. 能级组成:
运动状态 能级 电子运动 电子能级 振动运动 振动能级 转动运动 转动能级 + + - -

26
光吸收量子选律:分子(原子)吸收能量后,只能从一个能级跃迁到另一个能级。
吸收能量: 跃迁禁阻:分子(原子)在跃迁时,停留在两个能级之间的跃迁禁止发生。
在跃迁时,停留在两个能级之间的跃迁禁止发生。")
27
能级 能量差 对应波长 光谱区域 电子能级 1~20eV 0.06~1.25um 紫外、可见区 振动能级 0.05~1eV 1.25~25um 中红外区 转动能级 0.005~0.05eV 25~250um 远红外区 电子能级间隔比振动能级和转动能级间隔大1~2个数量级,在发生电子能级跃迁时,伴有振-转能级的跃迁,即电子光谱中总包含有振动能级和转动能级间跃迁产生的若干谱线,形成带状光谱。

28
不同物质结构不同导致分子能级的能量间隔各异,不同物质将选择性吸收不同波长的辐射,这是UV-Vis定性分析的基础。
定性分析是让不同波长的光通过待测物,测量其对不同波长光的吸收程度,以吸光度A为纵坐标,辐射波长为横坐标作图,得到该物质的吸收光谱,据吸收光谱的特性研究分子结构。

29
-胡罗卜素 咖啡因 几种有机化合物的分子吸收光谱图。 阿斯匹林 丙酮

30
吸收曲线反映物质对不同波长光的吸收能力。
二、分子吸收光谱——吸收曲线 分子结构的复杂性使其对不同波长光的吸收程度不同,用不同波长的单色光照射,测吸光度— 吸收曲线与最大吸收波长 max; 吸收曲线反映物质对不同波长光的吸收能力。

31
吸收曲线的讨论: (1)同一种物质对不同波长光的吸光度不同。吸光度最大处对应的波长称为最大吸收波长λmax
(2)不同浓度的同一种物质,其吸收曲线形状相似λmax不变。而对于不同物质,它们的吸收曲线形状和λmax则不同。 (3)吸收曲线可以提供物质的结构信息,并作为物质定性分析的依据之一。
不同浓度的同一种物质,其吸收曲线形状相似λmax不变。而对于不同物质,它们的吸收曲线形状和λmax则不同。 (3)吸收曲线可以提供物质的结构信息,并作为物质定性分析的依据之一。")
32
吸收曲线的讨论: (4)不同浓度的同一种物质,在某一定波长下吸光度 A 有差异,在λmax处吸光度A 的差异最大。此特性可作为物质定量分析的依据。 (5)在λmax处吸光度随浓度变化的幅度最大,所以测定最灵敏。吸收曲线是定量分析中选择入射光波长的重要依据。
不同浓度的同一种物质,在某一定波长下吸光度 A 有差异,在λmax处吸光度A 的差异最大。此特性可作为物质定量分析的依据。 (5)在λmax处吸光度随浓度变化的幅度最大,所以测定最灵敏。吸收曲线是定量分析中选择入射光波长的重要依据。")
33
讨论: (6)吸收光谱的波长分布是由产生谱带的跃迁能级间的能量差所决定,反映了分子内部能级分布状况,是物质定性的依据; (7)吸收谱带的强度与分子偶极矩变化、跃迁几率有关,也提供分子结构的信息。通常将在最大吸收波长处测得的摩尔吸光系数εmax也作为定性的依据。不同物质的λmax有时可能相同,但εmax不一定相同; (8)吸收谱带强度与该物质分子吸收的光子数成正比,定量分析的依据。
吸收谱带的强度与分子偶极矩变化、跃迁几率有关,也提供分子结构的信息。通常将在最大吸收波长处测得的摩尔吸光系数εmax也作为定性的依据。不同物质的λmax有时可能相同,但εmax不一定相同; (8)吸收谱带强度与该物质分子吸收的光子数成正比,定量分析的依据。")
34
三、吸收光谱主要类型 ultraviolet spectrometry of organic compounds
有机分子能级跃迁 有机分子轨道包括: 成键轨道 、 ; 反键轨道 *、* 非键轨道 n 例如 H2O分子的轨道 C H O o = = o=n

35
1.有机物的电子跃迁 C O H E n s * n p * s p p
R K E , B n p E C O H n p s 分子轨道理论:成键轨道—反键轨道。 当外层电子吸收紫外或可见辐射后,就从基态向激发态(反键轨道)跃迁。主要有四种跃迁所需能量ΔΕ大小顺序为: n →π* < π→π* < n →σ* < σ→σ*
跃迁。主要有四种跃迁所需能量ΔΕ大小顺序为: n →π* < π→π* < n →σ* < σ→σ*")
36
(1) σ→σ*跃迁 E n s * p * p 所需能量最大;σ电子只有吸收远紫外光的能量才能发生跃迁;
(1) σ→σ*跃迁 所需能量最大;σ电子只有吸收远紫外光的能量才能发生跃迁; 饱和烷烃的分子吸收光谱出现在远紫外区; 吸收波长λ<200 nm; 例:甲烷的λmax为125nm , 乙烷λmax为135nm。 只能被真空紫外分光光度计检测到; 作为溶剂使用; s p * s * R K E , B n p E
 σ→σ*跃迁. 所需能量最大;σ电子只有吸收远紫外光的能量才能发生跃迁; 饱和烷烃的分子吸收光谱出现在远紫外区; 吸收波长λ<200 nm; 例:甲烷的λmax为125nm , 乙烷λmax为135nm。 只能被真空紫外分光光度计检测到; 作为溶剂使用; s. p * s * R. K. E. , B. n. p. E.")
37
(2) n→σ*跃迁 所需能量较大。 吸收波长为150~250nm,大部分在远紫外区,近紫外区仍不易观察到。
含非键电子的饱和烃衍生物(含N、O、S和卤素等杂原子)均呈现n→σ* 跃迁。
均呈现n→σ* 跃迁。")
38
(3) π→π*跃迁 所需能量较小,吸收波长处于远紫外区的近紫外端或近紫外区,εmax一般在104L·mol-1·cm-1以上,属于强吸收。
①不饱和烃π→π*跃迁 乙烯π→π*跃迁的λmax为162nm,εmax为:1×104L·mol-1·cm-1。 K带——共轭非封闭体系的p → p* 跃迁,强带吸收 C=C 发色基团, 但 → *一般在200nm附近。 max=162nm 助色基团取代 (K带)发生红移。
发生红移。")
39
基-----是由非环或六环共轭二烯母体决定的基准值;
②共轭烯烃中的 → * 165nm 217nm ₃ ₁ ₂ (HOMO LVMO) max 共轭烯烃(不多于四个双键) *跃迁吸收峰位置可由伍德沃德——菲泽 规则估算。 max= 基+nii 基-----是由非环或六环共轭二烯母体决定的基准值; 无环、非稠环二烯母体: 基=217 nm
 max 共轭烯烃(不多于四个双键) *跃迁吸收峰位置可由伍德沃德——菲泽 规则估算。 max= 基+nii. 基-----是由非环或六环共轭二烯母体决定的基准值; 无环、非稠环二烯母体: 基=217 nm.")
40
niI : 由双键上取代基种类和个数决定的校正项
异环(稠环)二烯母体: 基=214 nm 同环(非稠环或稠环)二烯母体: 基=253 nm niI : 由双键上取代基种类和个数决定的校正项 (1)每增加一个共轭双键 +30 (2)环外双键 (3)双键上取代基: 酰基(-OCOR) 卤素(-Cl,-Br) +5 烷基(-R) 烷氧基(-OR)
二烯母体: 基=214 nm. 同环(非稠环或稠环)二烯母体: 基=253 nm. niI : 由双键上取代基种类和个数决定的校正项. (1)每增加一个共轭双键 +30. (2)环外双键 +5. (3)双键上取代基: 酰基(-OCOR) 0 卤素(-Cl,-Br) +5. 烷基(-R) +5 烷氧基(-OR) +6.")
41
③羰基化合物共轭烯烃中的 → * K R n Ⅰ Y=H,R n → * 180-190nm
③羰基化合物共轭烯烃中的 → * K R n Ⅰ Y=H,R n → * nm → * nm n → * nm Ⅱ Y= -NH2,-OH,-OR 等助色基团 K 带红移,R 带兰移; R带max =205nm ;10-100 n 165nm Ⅲ 不饱和醛酮 K带红移:165250nm R 带蓝移:290310nm

42
④芳香烃及其杂环化合物 苯: E1带180184nm; =47000 E2带200204 nm =7000
苯环上三个共扼双键的 → *跃迁特征吸收带; B带 nm =200 → *与苯环振动引起; 含取代基时, B带简化,红移。 max(nm) max 苯 254 200 甲苯 261 300 间二甲苯 263 1,3,5-三甲苯 266 305 六甲苯 272
 max. 苯 甲苯 间二甲苯 ,3,5-三甲苯 六甲苯")
43
乙酰苯紫外光谱图 n→ p* ; R带 p → p* ; K带 羰基双键与苯环共扼: K带强;苯的E2带与K带合并,红移;
取代基使B带简化; 氧上的孤对电子: R带,跃迁禁阻,弱; C H 3 O n→ p* ; R带 p → p* ; K带

44
苯环上助色基团对吸收带的影响

45
苯环上发色基团对吸收带的影响

46
⑤立体结构和互变结构的影响 顺反异构: 互变异构: 顺式:λmax=280nm; εmax=10500

47
立体结构和互变结构的影响

48
(4) n → *跃迁 所需能量最小,吸收波长处于近紫外区到可见光区,εmax一般在100左右,属于弱吸收,称为R带。
 n → *跃迁 所需能量最小,吸收波长处于近紫外区到可见光区,εmax一般在100左右,属于弱吸收,称为R带。")
49
2. 溶剂的影响 n p 非极性 极性 n >p n<p n n p
非极性 极性 n >p n<p n n p 非极性 极性 n → *跃迁:蓝移; ; → *跃迁:红移; ; max(正己烷) max(氯仿) max(甲醇) max(水) * 230 238 237 243 n* 329 315 309 305
 max(氯仿) max(甲醇) max(水) * n*")
50
溶剂的影响 极性溶剂使精细结构消失; 1:乙醚 2:水 1 2 250 300 苯酰丙酮 非极性 → 极性
n → *跃迁:兰移; ; → *跃迁:红移; ; 极性溶剂使精细结构消失;

51
3.生色团与助色团 生色团:π→π*和n→π*两种跃迁均要求有机物分子中含有不饱和基团。这类含有π键的不饱和基团称为生色团。简单的生色团由双键或叁键体系组成,如乙烯基、羰基、亚硝基、偶氮基-N=N-、乙炔基、腈基-C≡N等。 助色团:有一些含有n电子的基团(如-OH、-OR、-NH2、-NHR、-X等),它们本身没有生色功能(不能吸收λ>200nm的光),但当它们与生色团相连时,就会发生n-π共轭作用,增强生色团的生色能力(吸收波长向长波方向移动,且吸收强度增加),这样的基团称为助色团。 常见助色团助色顺序:-F<-CH3<-Br<-OH<-OCH3<-NH2<-NHCH3<-NH(CH3)2<-NHC6H5<-O-
,它们本身没有生色功能(不能吸收λ>200nm的光),但当它们与生色团相连时,就会发生n-π共轭作用,增强生色团的生色能力(吸收波长向长波方向移动,且吸收强度增加),这样的基团称为助色团。 常见助色团助色顺序:-F<-CH3<-Br<-OH<-OCH3<-NH2<-NHCH3<-NH(CH3)2<-NHC6H5<-O-")
52
增色和减色效应 有机化合物的吸收谱带常常因引入取代基或改变溶剂使最大吸收波长λmax和吸收强度发生变化:

53
§4 光的吸收基本定律 ──朗伯-比耳(Lambert-Beer)定律
一、朗伯—比耳定律 一束平行单色光通过溶液: I0=Ia+It+Ir

54
Lambert定律: Beer定律: lgI0/It∝b lgI0/It ∝ c 朗伯-比耳定律:lgI0/It ∝ b c

55
朗伯—比耳定律数学表达式 式中 A:吸光度;描述溶液对光的吸收程度; b:液层厚度(光程长度),通常以cm为单位; c:溶液的浓度;
K:吸光系数 摩尔吸光系数:ε 单位: L·mol-1·cm-1 吸光系数:a 单位: L·g-1·cm-1 关系: a = ε / M

56
透光度T 透光度T : 描述入射光透过溶液的程度 T = It / I0 吸光度A与透射比T 的关系: (1) 吸光光度法的理论基础和定量测定的依据。 (2) 摩尔吸收系数 ε 在数值上等于浓度为1 mol ·L-1、液层厚度为1cm时该溶液在某一波长下的吸光度; (3) 吸光度具有加和性。
 吸光度具有加和性。")
57
摩尔吸光系数ε的讨论 (1)吸收物质在一定波长和溶剂条件下的特征常数;
(2)不随浓度c和光程长度b的改变而改变。在温度和波长等条件一定时, ε仅与吸收物质本身的性质有关,与待测物浓度无关; (3)同一吸收物质在不同波长下的ε值是不同的。在最大吸收波长λmax处的摩尔吸光系数ε max表明了该吸收物质最大限度的吸光能力,也反映了光度法测定该物质可能达到的最大灵敏度。
不随浓度c和光程长度b的改变而改变。在温度和波长等条件一定时, ε仅与吸收物质本身的性质有关,与待测物浓度无关; (3)同一吸收物质在不同波长下的ε值是不同的。在最大吸收波长λmax处的摩尔吸光系数ε max表明了该吸收物质最大限度的吸光能力,也反映了光度法测定该物质可能达到的最大灵敏度。")
58
摩尔吸收系数ε的讨论 (4)可作为定性鉴定的参数; (5)物质的吸光能力的度量 ε = (6~10)×104 :高灵敏;
ε max越大表明该物质的吸光能力越强,用光度法测定该物质的灵敏度越高。 ε >105:超高灵敏; ε = (6~10)×104 :高灵敏; ε = 104~ 103 :中等灵敏; ε < :不灵敏。 (6) ε在数值上等于浓度为1 mol ·L-1、液层厚度为1cm时该溶液在某一波长下的吸光度。
×104 :高灵敏; ε = 104~ 103 :中等灵敏; ε < 103 :不灵敏。 (6) ε在数值上等于浓度为1 mol ·L-1、液层厚度为1cm时该溶液在某一波长下的吸光度。")
59
二、影响光吸收定律的因素 A = K b c 1. 现象 标准曲线法测定未知溶液的浓度时,发现:标准曲线常发生弯曲(尤其当溶液浓度较高时),这种现象称为对朗伯—比耳定律的偏离。 2. 引起偏离的因素(两大类) (1)物理性因素, 即仪器的非理想引起的; (2)化学性因素。
物理性因素, 即仪器的非理想引起的; (2)化学性因素。")
60
物理性因素: 难以获得真正的纯单色光。 分光光度计只能获得近乎单色的狭窄光带。复合光可导致对朗伯—比耳定律的正或负偏离。
非单色光、杂散光、非平行入射光都会引起对朗伯—比耳定律的偏离,最主要的是非单色光作为入射光引起的偏离。

61
非单色光作为入射光引起的偏离: 假设由波长为λ1和λ2的两单色光 组成的入射光通过浓度为c的溶液,则: A1=lg(I01 /It1 )= ε 1 bc A2=lg(I02 /It2 )= ε 2 bc 故: 式中:I01、I02分别为λ1、λ2 的入射光强度; It1 、It2分别为λ1、λ2 的透射光强度; ε 1、 ε 2分别为λ1、λ2的摩尔吸收系数;

62
因实际上只能测总吸光度A总,故 A总 = lg(I0总/It总 ) = lg(I01 +I02)/(It1 +It2 ) = lg(I01 +Io2)/(I ε 1bc +I ε 2bc ) 令: ε 1 - ε 2 = ε ; 设: I01 =I02 A总 = lg(2I01)/It1( ε bc ) = A 1 + lg2 - lg( ε bc )
/It1( ε bc ) = A 1 + lg2 - lg( ε bc )")
63
讨论: A总 =A1 + lg2 - lg(1+10- ε bc ) (1) = 0; 即: ε 1= ε 2 = ε 则: A总 =lg(Io /It)= ε bc (2) ε ≠0 若 ε <0 ;即ε 2< ε 1 ; - ε bc>0, lg(1+10 εbc )值随c值增大而增大,则标准曲线偏离直线向c 轴弯曲,即负偏离; 反之,则向A轴弯曲,即正偏离。
值随c值增大而增大,则标准曲线偏离直线向c 轴弯曲,即负偏离; 反之,则向A轴弯曲,即正偏离。")
64
(4) 为克服非单色光引起的偏离,首先应选择比较好的单色器。此外还应将入射波长选定在待测物质的最大吸收波长且吸收曲线较平坦处。
讨论: (3) | ε |很小时,即ε 1≈ ε 2: 可近似认为是单色光。在低浓度范围内,不发生偏离。若浓度较高,即使| ε |很小, A总 ≠A1 ,且随着c值增大, A总 与A 1的差异愈大,表现为A-c曲线上部(高浓度区)弯曲愈严重。故朗伯-比耳定律只适用于稀溶液。 (4) 为克服非单色光引起的偏离,首先应选择比较好的单色器。此外还应将入射波长选定在待测物质的最大吸收波长且吸收曲线较平坦处。
 | ε |很小时,即ε 1≈ ε 2: 可近似认为是单色光。在低浓度范围内,不发生偏离。若浓度较高,即使| ε |很小, A总 ≠A1 ,且随着c值增大, A总 与A 1的差异愈大,表现为A-c曲线上部(高浓度区)弯曲愈严重。故朗伯-比耳定律只适用于稀溶液。 (4) 为克服非单色光引起的偏离,首先应选择比较好的单色器。此外还应将入射波长选定在待测物质的最大吸收波长且吸收曲线较平坦处。")
65
讨论: 选用谱带a的复合光进行测量,得到右图的工作曲线,A与c基本呈直线关系。 选用谱带b的复合光进行测量, κ的变化较大, 则A随波长的变化较明显,得到的工作曲线明显偏离线性。

66
(2) 化学性因素 朗伯-比耳定律的假定:所有的吸光质点之间不发生相互作用;仅在稀溶液(c<10-2 mol ·L-1)时才基本符合
故:朗伯-比耳定律只适用于稀溶液。 溶液中存在着离解、聚合、互变异构、配合物的形成等化学平衡时。使吸光质点的浓度发生变化。 例: CrO H+ = Cr2O72- + H2O

67
§5 分光光度计

68
分光光度计

69
一、仪器结构 光源 分光系统 样品室 检测器 显示 (数据处理系统)
")
70
紫外区:氢、氘灯。发射150~400 nm的连续光谱,使用波长190~350nm。
1. 光源 要求:在整个紫外光区或可见光区可以发射连续光谱,具有足够的辐射强度、较好的稳定性、较长的使用寿命。 可见光区:钨灯、卤钨灯。辐射波长范围在320~2500 nm,使用波长350~1100nm。 紫外区:氢、氘灯。发射150~400 nm的连续光谱,使用波长190~350nm。

71
2.分光系统 单色器:获得高光谱纯度辐射束的装置,而辐射束的波长可在很宽范围内任意改变; 主要部件: (1)进口狭缝;
(2)准直装置(透镜或反射镜):使辐射束成为平行光线; (3)色散装置(棱镜、光栅):使不同波长的辐射以不同的角度进行传播;
准直装置(透镜或反射镜):使辐射束成为平行光线; (3)色散装置(棱镜、光栅):使不同波长的辐射以不同的角度进行传播;")
72
(4)聚焦透镜或凹面反射镜,使每个单色光束在单色器的出口曲面上成像。
聚焦透镜或凹面反射镜,使每个单色光束在单色器的出口曲面上成像。")
73
棱镜 平行光经过棱镜后按波长顺序排列成为单色光;经聚焦后在焦面上的不同位置上成像,获得按波长展开的光谱;
棱镜对不同波长的光具有不同的折射率,波长长的光,折射率小;波长短的光,折射率大。 平行光经过棱镜后按波长顺序排列成为单色光;经聚焦后在焦面上的不同位置上成像,获得按波长展开的光谱; 棱镜的分辨能力取决于棱镜的几何尺寸和材料; 棱镜的光学特性可用色散率和分辨率来表征;

74
棱镜的特性与参数 (1)色散率 角色散率:用dθ/dλ表示,偏向角θ对波长的变化率; 倒线色散率:用dλ/dl 表示,
棱镜的顶角越大或折射率越大,角色散率越大,分开两条相邻谱线的能力越强,但顶角越大,反射损失也增大,通常为60度角; 线色散率:用dl /dλ表示,两条相邻谱线在焦面上被分开的距离对波长的变化率; 倒线色散率:用dλ/dl 表示,

75
(2)分辨率 相邻两条谱线分开程度的度量: :两条相邻谱线的平均波长;△λ:两条谱线的波长差;
b:棱镜的底边长度;n:棱镜介质材料的折射率。 分辨率与波长有关,长波的分辨率要比短波的分辨率小,棱镜分离后的光谱属于非均排光谱。

76
光栅 透射光栅,反射光栅; 光栅光谱的产生是多狭缝干涉与单狭缝衍射共同作用的结果,前者决定光谱出现的位置,后者决定谱线强度分布;

77
光栅的特性: 图所示反射光栅是由与光栅表面成β角的小斜面构成(小阶梯光栅,闪耀光栅),β角叫做闪耀角。
将反射光栅的线槽加工成适当形状能使有效强度集中在特定的衍射角上。 图所示反射光栅是由与光栅表面成β角的小斜面构成(小阶梯光栅,闪耀光栅),β角叫做闪耀角。 选择适宜的闪耀角,可以使90%的有效能量集中在单独一级的衍射上。
,β角叫做闪耀角。 选择适宜的闪耀角,可以使90%的有效能量集中在单独一级的衍射上。")
78
光栅的参数: 角色散率只与色散元件的性能有关;线色散率还与仪器的焦距有关。
光栅的特性可用色散率和分辨率来表征,当入射角不变时,光栅的角色散率可通过对光栅公式求导得到: dθ/dλ为入射角对波长的变化率,即光栅的角色散率。 当θ很小,且变化不大时,cosθ ≈1,光栅的角色散率决定于光栅常数 d 和光谱级数n ,常数,不随波长改变,均排光谱(优于棱镜)。 角色散率只与色散元件的性能有关;线色散率还与仪器的焦距有关。
。 角色散率只与色散元件的性能有关;线色散率还与仪器的焦距有关。")
79
光栅的分辨率R 光栅的分辨率R 等于光谱级次(n)与光栅刻痕条数(N)的乘积: 光栅越宽、单位刻痕数越多、R 越大。
宽度50mm,N=1200条/mm, 一级光谱的分辨率: R=1×50×1200=6×104

80
狭 缝 单色器的进口狭缝起着单色器光学系统虚光源的作用。复合光经色散元件分开后,在出口曲面上形成相当于每条光谱线的像,即光谱。转动色散元件可使不同波长的光谱线依次通过。 分辨率大小不仅与色散元件的性能有关,也取决于成像的大小,因此希望采用较窄的进口狭缝。分辨率用来衡量单色器能分开波长的最小间隔的能力;最小间隔的大小用有效带宽表示: S = DW D为线色散率的倒数;W为狭缝宽度;

81
定性分析时,减小狭缝宽度,使相邻谱线的分辨率提高;
在光谱分析中: 定性分析时,减小狭缝宽度,使相邻谱线的分辨率提高; 定量分析时,增大狭缝宽度,可使光强增加。 狭缝两边的边缘应锐利且位于同一平面上;

82
3. 样品池 样品池放置各种类型的吸收池(比色皿)和相应的池架附件。吸收池主要有石英池和玻璃池两种。在紫外区须采用石英池,可见光区一般用玻璃池。
和相应的池架附件。吸收池主要有石英池和玻璃池两种。在紫外区须采用石英池,可见光区一般用玻璃池。")
83
4. 检测系统 将光强度转换成电流来进行测量。光电检测器。 要求:对测定波长范围内的光有快速、灵敏的响应,
产生的光电流应与照射于检测器上的光强度成正比。 (1)光电倍增管 优点:高灵敏度;响应快;适于弱光测定,甚至对单一光子均可响应。 缺点:热发射强,因此暗电流大,需冷却(-30oC)。不得置于强光(如 日光)下,否则可永久损坏 PMT!
光电倍增管. 优点:高灵敏度;响应快;适于弱光测定,甚至对单一光子均可响应。 缺点:热发射强,因此暗电流大,需冷却(-30oC)。不得置于强光(如. 日光)下,否则可永久损坏 PMT!")
84
光电倍增管(photomultiplier tube, PMT) 共有9个倍增阴极(dynatron),所加直流电压共为9010V
石英套 光束 1个光子产生106~107个电子 栅极,Grill 阳极 屏蔽 光电倍增管示意图 共有9个倍增阴极(dynatron),所加直流电压共为9010V
,所加直流电压共为9010V.")
85
(2)光电二极管阵列检测器 电容器 充电 放电 光照射 再次充电 测量周期
电容器再次充电的电量与每个二极管检测到的光子数目成正比,而光子数又与光强成正比。通过测量整个波长范围内光强的变化就可得到吸收光谱。

86
5. 数据处理系统 计算机工作站 作用:数据显示,数据处理

87
二、分光光度计的类型 types of spectrometer
1.单光束 简单,价廉,适于在给定波长处测量吸光度或透光度,一般不能作全波段光谱扫描,要求光源和检测器具有很高的稳定性。 2.双光束 自动记录,快速全波段扫描。可消除光源不稳定、检测器灵敏度变化等因素的影响,特别适合于结构分析。仪器复杂,价格较高。

88
3.双波长 将不同波长的两束单色光(λ1、λ2) 快束交替通过同一吸收池而后到达检测器。产生交流信号。无需参比池。△= 1~2nm。两波长同时扫描即可获得导数光谱。
 快束交替通过同一吸收池而后到达检测器。产生交流信号。无需参比池。△= 1~2nm。两波长同时扫描即可获得导数光谱。")
90
4. 光学性能 波长范围:测量范围190~900nm 波长准确度:≤±0.05nm 波长重现性:≤0.05nm 透光率测量范围:0~150% 吸光度测量范围:-0.173~+2.00 光度准确度:透光率满量程误差≤±0.5% 光度重复性:透光率变动性≤±0.5% 分辨率:260nm处⊿λ=0.3nm 杂散光:220nm处NaI(1% g/ml) ≤0.5%
 ≤0.5%")
91
5. 分光光度计的校正 波长标度校正: 使用镨-钕玻璃(可见光区)和钬玻璃(紫外光区)进行校正。因为二者均有其各自的特征吸收峰。 吸光度标度校正: 采用 K2CrO4 标准液校正,在25oC时,于不同波长处测定 g/L的 KOH 溶液(0.05mol/L)的吸光度 A,调整光度计使其A 达到规定的吸光度。 吸收池的校正(配对): 两个吸收池吸光度测量差值应小于1%
的吸光度 A,调整光度计使其A 达到规定的吸光度。 吸收池的校正(配对): 两个吸收池吸光度测量差值应小于1%")
92
§6 反应条件和测量条件的选择 一、显色反应及条件的选择 1.选择显色反应时,应考虑的因素 2.配位显色反应
§6 反应条件和测量条件的选择 一、显色反应及条件的选择 1.选择显色反应时,应考虑的因素 灵敏度高、选择性高、生成物稳定、显色剂在测定波长处无明显吸收,两种有色物最大吸收波长之差:“对比度”,要求△ > 60 nm。 2.配位显色反应 当金属离子与有机显色剂形成配合物时,通常会发生电荷转移跃迁,产生很强的紫外—可见吸收光谱。

93
(一)显色反应和显色剂 1.显色反应 在分光光度分析中,将试样中被测组分转变成有色化合物的反应叫显色反应。 显色反应可分配位反应、耦合反应和氧化还原反应。与被测组分化合成有色物质的试剂称为显色剂。 同一组分可与若干种显色剂反应,生成若干有色化合物,其原理和灵敏度亦有差别。一种被测组分究竞应该用哪种显色反应,可根据所需标准加以选择。

94
选择显色反应的一般要求 : (1)待测组分应定量转变成有色化合物,二者有确定的化学计量关系。
(2)灵敏度高。灵敏度的高低可从摩尔吸光系数值的大小来判断,ε值大灵敏度高,否则灵敏度低。但灵敏度高的显色反应,不一定选择性就好,对于高含量的组分不一定要选用灵敏度高的显色反应。 (3)对比度要大。即如果显色剂有颜色,则有色化合物与显色剂的最大吸收波长的差别要大,一般要求⊿λ在60nm以上。
灵敏度高。灵敏度的高低可从摩尔吸光系数值的大小来判断,ε值大灵敏度高,否则灵敏度低。但灵敏度高的显色反应,不一定选择性就好,对于高含量的组分不一定要选用灵敏度高的显色反应。 (3)对比度要大。即如果显色剂有颜色,则有色化合物与显色剂的最大吸收波长的差别要大,一般要求⊿λ在60nm以上。")
95
(4)有色化合物的组成恒定,化学性质稳定。有色化合物的组成若不确定,测定的再现性就较差。有色化合物若易受空气的氧化、日光的照射而分解,就会引入测量误差。
(5)显色反应的条件要易于控制。如果条件要求过于严格,难以控制,测定结果的再现性就差。 (6)选择性好,干扰少,或者干扰离子易被消除、或者显色剂与被测组分和干扰离子生成的有色化合物的吸收峰相隔较远。
显色反应的条件要易于控制。如果条件要求过于严格,难以控制,测定结果的再现性就差。 (6)选择性好,干扰少,或者干扰离子易被消除、或者显色剂与被测组分和干扰离子生成的有色化合物的吸收峰相隔较远。")
96
2.显色剂 (1)无机显色剂 许多无机试剂能与金属离子起显色反应。但是多数无机显色剂的灵敏度和选择性都不高,其中性能较好,目前还有实用价值的有硫氰酸盐、钼酸铵、氨水和过氧化氢等。 (2)有机显色剂 许多有机试剂,在一定条件下,能与金属离子生成有色的金属螯合物(具有环状结构的络合物)。 偶氮类显色剂:性质稳定、显色反应灵敏度高、选择性好、对比度大,应用最广泛。偶氮胂III、PAR等。 三苯甲烷类:铬天青S、二甲酚橙等
。 偶氮类显色剂:性质稳定、显色反应灵敏度高、选择性好、对比度大,应用最广泛。偶氮胂III、PAR等。 三苯甲烷类:铬天青S、二甲酚橙等.")
97
1.大部分金属螯合物都呈现鲜明的颜色,ε大于104,因而测定的灵敏度很高; 2.金属螯合物都很稳定,一般离解常数都很小,而且能抗辐射;
金属螯合物用于光度分析的优点: 1.大部分金属螯合物都呈现鲜明的颜色,ε大于104,因而测定的灵敏度很高; 2.金属螯合物都很稳定,一般离解常数都很小,而且能抗辐射; 3.专用性强,绝大多数有机螯合剂在一定条件下,只与少数或一种金属离子络合,且同一种有机螯合剂与不同的金属离子络合时,生成具有特征颜色的螯合物。

98
4.大部分金属螯合物难溶于水,但可被萃取到有机溶剂中,发展了萃取光度法。
5.在显色分子中,金属所占的比率很低,提高了测定的灵敏度。因此,有机显色剂是光度分析中应用最多最广的显色剂,寻找高选择性、高灵敏度的有机显色剂,是光度分析发展和研究的重要内容。

99
含有生色基团的有机化合物常常能与许多全属离子化合生成性质稳定且具有特征颜色的化合物,且灵敏度和选择性都很高,这就为用光度法测定这些离子提供了很好的条件。
常用有机显色剂: 1. 邻二氮菲 2. 双硫腙 3. 二甲酚橙 4. 偶氮胂III 5. 铬天青S

100
(二)显色反应条件的选择 1.显色剂的用量 显色就是将被测组分转变成有色化合物,表示: M R = MR (被测组分) (显色剂) (有色化合物) 反应在一定程度上是可逆的。为了减少反应的可逆性,根据同离子效应,加入过量的显色剂是必要的,但也不能过量太多,否则会引起副反应,对测定反而不利。

101
合适的显色剂用量,应通过实验确定 单因素法

102
2.溶液酸度 溶液酸度直接影响着金属离子和显色剂的存在形式以及有色络合物的组成和稳定性。 (1)酸度对被测物质存在状态的影响 大部分高价金属离子都易水解,溶液的酸度降低时,会产生一系列羟基络离子或多核羟基络离子。高价金属离子的水解分级进行。 随着水解的进行,同时还发生各种类型的聚合反应。聚合度随着时间增大,最终将导致沉淀的生成。

103
(2) 酸度对显色剂浓度和颜色的影响 大部分显色剂都是有机弱酸。显色反应进行时,首先是有机弱酸发生离解,其次才是络阴离子与金属离子络合。 M + HR=MR H+ 溶液的酸度影响着显色剂的离解,并影响着显色反应的完全程度。
 酸度对显色剂浓度和颜色的影响 大部分显色剂都是有机弱酸。显色反应进行时,首先是有机弱酸发生离解,其次才是络阴离子与金属离子络合。 M + HR=MR + H+ 溶液的酸度影响着显色剂的离解,并影响着显色反应的完全程度。")
104
许多显色剂本身就是酸碱指示剂,当溶液酸度改变时,显色剂本身就有颜色变化。如果显色剂在某一酸度时,配位反应和指示剂反应同时发生,两种颜色同时存在,就无法进行光度测定。
如二甲酚橙在溶液的pH>6.3时呈红色,在pH<6.3时呈柠檬黄色,在pH=6.3时,呈中间色。而二甲酚橙与金属离子的络合物呈现红色。故二甲酚橙只有在pH<6的酸性溶液中可作为金属离子显色剂。

105
(3)对配合物组成和颜色的影响 对于某些逐级形成络合物的显色反应、在不同酸度时,生成不同配位比的配合物。例如铁与水杨酸的络合反应, pH< [Fe3+(C7H4O3)2-]+ 紫色 4<pH<9 [Fe3+(C7H4O3)22-]- 红色 pH> [Fe3+(C7H4O3)32-]3- 黄色 在这种情况下,必须控制合适的酸度,才可获得好的分析结果。
![(3)对配合物组成和颜色的影响 对于某些逐级形成络合物的显色反应、在不同酸度时,生成不同配位比的配合物。例如铁与水杨酸的络合反应, pH<4 [Fe3+(C7H4O3)2-]+ 紫色.](http://slidesplayer.com/slide/11394544/61/images/105/%283%29%E5%AF%B9%E9%85%8D%E5%90%88%E7%89%A9%E7%BB%84%E6%88%90%E5%92%8C%E9%A2%9C%E8%89%B2%E7%9A%84%E5%BD%B1%E5%93%8D+%E5%AF%B9%E4%BA%8E%E6%9F%90%E4%BA%9B%E9%80%90%E7%BA%A7%E5%BD%A2%E6%88%90%E7%BB%9C%E5%90%88%E7%89%A9%E7%9A%84%E6%98%BE%E8%89%B2%E5%8F%8D%E5%BA%94%E3%80%81%E5%9C%A8%E4%B8%8D%E5%90%8C%E9%85%B8%E5%BA%A6%E6%97%B6%EF%BC%8C%E7%94%9F%E6%88%90%E4%B8%8D%E5%90%8C%E9%85%8D%E4%BD%8D%E6%AF%94%E7%9A%84%E9%85%8D%E5%90%88%E7%89%A9%E3%80%82%E4%BE%8B%E5%A6%82%E9%93%81%E4%B8%8E%E6%B0%B4%E6%9D%A8%E9%85%B8%E7%9A%84%E7%BB%9C%E5%90%88%E5%8F%8D%E5%BA%94%EF%BC%8C+pH%EF%BC%9C4+%5BFe3%2B%28C7H4O3%292-%5D%2B+%E7%B4%AB%E8%89%B2..jpg "4<pH<9 [Fe3+(C7H4O3)22-]- 红色. pH>9 [Fe3+(C7H4O3)32-]3- 黄色. 在这种情况下,必须控制合适的酸度,才可获得好的分析结果。")
106
3. 显色温度 一般显色反应可在室温下完成。但有些显色反应需要加热至一定的温度才能完成;也有些有色配合物在较高温度下容易分解。 应根据不同的情况选择适当的温度进行显色。温度对光的吸收及颜色的深浅也有一定的影响,故标样和试样的显色温度应保持一样。合适显色温度也必须通过实验确定,做A-T曲线求出。

107
4. 显色时间 大多数显色反应速度较慢,需要一定时间,溶液的颜色才能达到稳定程度。有些有色化合物放置一段时间后,由于空气的氧化,试剂的分解或挥发,光的照射等原因,使颜色减退。 适宜的显色时间和有色溶液稳定程度,必须通过实验来确定。绘制A-t曲线,根据曲线来确定适宜的时间。

108
5.有机溶剂和表面活性剂 (1)溶剂影响配合物的离解度:许多有色化合物在水中的离解度大,而在有机溶剂中的离解度小,如在Fe(SCN)3溶液中加入可与水混溶的有机试剂(如丙酮),降低Fe(SCN)3的离解度,使颜色加深,提高了测定的灵酸度。 (2)溶剂改变配合物颜色:各种溶剂分子的极性不同、介电常数不同,从而影响到配合物的稳定性,改变了配合物分子内部的状态或者形成不同的溶剂化物的结果。
溶剂改变配合物颜色:各种溶剂分子的极性不同、介电常数不同,从而影响到配合物的稳定性,改变了配合物分子内部的状态或者形成不同的溶剂化物的结果。")
109
(3)溶剂影响显色反应的速度 当用氯代磺酚S测定Nb时,在水溶液中显色需几小时,加入丙酮后,仅需30分钟。 常用纯溶剂的截止波长: 水190nm、乙腈190nm、甲醇205nm、 正己烷210nm、乙醚220nm、 四氢呋喃225nm、二氯甲烷245nm、 氯仿245nm。

110
表面活性剂: 表面活性剂的加入可以提高显色反应的灵敏度,增加有色化合物的稳定性。其作用原理一方面是胶束增溶,另一方面是可形成含有表面活性剂的多元络合物。

111
6.共存离子的干扰及消除 (1) 与试剂生成有色络合物。如用硅钼蓝光度法测定钢中硅时,磷也能与钼酸铵生成络合物,同时被还原为钼蓝,使结果偏高。 (2)干扰离子本身有颜色。如Co2+(红色)、Cr3+(绿色)、Cu2+(蓝色)。 (3)与试剂结合成无色络合物消耗大量试剂而使被测离子络合不完全。如用水扬酸测Fe3+时,Al3+、Cu2+等有影响。 (4)与被测离子结合成离解度小的另一化合物。如由于F-的存在,能与Fe3+以FeF63-形式存在,Fe(SCN)3本不会生成,因而无法进行测定。
与试剂结合成无色络合物消耗大量试剂而使被测离子络合不完全。如用水扬酸测Fe3+时,Al3+、Cu2+等有影响。 (4)与被测离子结合成离解度小的另一化合物。如由于F-的存在,能与Fe3+以FeF63-形式存在,Fe(SCN)3本不会生成,因而无法进行测定。")
112
消除干扰的方法: (1)控制酸度 许多显色剂是有机弱酸,控制溶液的酸度,就可以控制显色剂R的浓度,就可以使某种金属离子显色,使另外一些金属离子不能生成有色络合物。 当溶液的情况比较复杂,或各种常数值不知道队则溶液最适合的pH值须通过实验方法来确定。

113
(2)加入掩蔽剂 在显色溶液里加一种能与干扰离子反应生成无色配合物的试剂。如用硫氰酸盐作显色剂测定Co2+,Fe3+有干优可加入氟化物,使Fe3+与F-结合生成无色而稳定的FeF63-,就可以消除干扰。 (3)采用萃取光度法 用适当的有机溶剂萃取有色组分,如用丁二酮肟测定钯时,钯与丁二酮肟所形成的内络盐,可被氯仿从酸性溶液中选择性地萃取。干扰离子不被萃取。
采用萃取光度法 用适当的有机溶剂萃取有色组分,如用丁二酮肟测定钯时,钯与丁二酮肟所形成的内络盐,可被氯仿从酸性溶液中选择性地萃取。干扰离子不被萃取。")
114
(4)在不同波长下测定两种显色配合物的吸光度,对它们进行同时测定。
(5)寻找新的显色反应 如将二元配合物改变为三元配合物。 (6) 分离干扰离子 在没有适当掩蔽剂时,干扰离于可用电解法,淀法或离子交换法等分离除去。 此外,还可以通过选择适当的测量条件,消除干扰离子的影响。
寻找新的显色反应. 如将二元配合物改变为三元配合物。 (6) 分离干扰离子. 在没有适当掩蔽剂时,干扰离于可用电解法,淀法或离子交换法等分离除去。 此外,还可以通过选择适当的测量条件,消除干扰离子的影响。")
115
二、吸光度测量条件的选择 1. 选择适当的测量波长 如图选500nm波长测定,灵敏度虽有所下降,却消除了干扰,提高了测定的准确度和选择性。
一般应该选择λmax为测量波长。 如果λmax处有共存组分干扰时,则应考虑选择灵敏度稍低但能避免干扰的入射光波长。 如图选500nm波长测定,灵敏度虽有所下降,却消除了干扰,提高了测定的准确度和选择性。 选择原则:吸收大,干扰小

116
2. 吸光度读数范围的选择 不同的透射比读数,产生的误差大小不同: -lgT= Kbc
微分:-dlgT=-0.434dlnT = T -1 dT = Kb dc 两式相除得: Δc/c不仅与仪器的透射比误差ΔT 有关,而且与其透射比读数T 的值也有关。 是否存在最佳读数范围?何值时误差最小?

117
最佳读数范围与最佳值: 设:ΔT =1%, Δc/c ~T 关系曲线 T 在15%~65% (A=0.2~0.8)之间时, 浓度相对误差较小, 最佳读数范围 浓度相对误差最小时的透射比Tmin为: Tmin= 36.8%, Amin= 0.434 通过改变吸收池厚度或待测液浓度,使吸光度读数处在适宜范围。普通分光光度法不适于高含量或极低含量物质的测定。

118
3. 参比溶液的选择 为什么需要使用参比溶液? 选择空白溶液的一般原则:
先用参比溶液调节透光度为100%,测得的的吸光度真正反映待测溶液吸光强度。 选择空白溶液的一般原则: ⑴ 溶剂空白:若仅待测组分与显色剂反应产物在测定波长处有吸收,其他所加试剂均无吸收,用纯溶剂(水)作参比溶液;
作参比溶液;")
119
⑵ 试剂空白:若显色剂或其他所加试剂在测定波长处略有吸收,而试液本身无吸收,用“试剂空白”(不加试样溶液)作参比溶液;
⑶ 试样空白:若待测试液在测定波长处有吸收,而显色剂等无吸收,则可用“试样空白”(不加显色剂)作参比溶液; ⑷ 平行操作空白:若显色剂、试液中其他组分在测量波长处有吸收,则可在试液中加入适当掩蔽剂将待测组分掩蔽后再加显色剂,作为参比溶液。
作参比溶液; ⑷ 平行操作空白:若显色剂、试液中其他组分在测量波长处有吸收,则可在试液中加入适当掩蔽剂将待测组分掩蔽后再加显色剂,作为参比溶液。")
121
第七节 定性及定量 分析方法 一、 定性分析 qualitative analysis 二、纯度检测 三、定量分析
quanti-tative analysis 四、有机物结构确定 structure determination of organic compounds 第七节 定性及定量 分析方法

122
一、定性分析 max:化合物特性参数,可作为定性依据; max , max都相同,可能是一个化合物;
有机化合物紫外吸收光谱:反映结构中生色团和助色团的特性,不完全反映分子特性; 计算吸收峰波长,确定共扼体系等 甲苯与乙苯:谱图基本相同; 结构确定的辅助工具; max , max都相同,可能是一个化合物; 标准谱图库:46000种化合物紫外光谱的标准谱图 «The sadtler standard spectra ,Ultraviolet»

123
二、纯度检测 根据吸收曲线的特征可检查样品中有无杂质。如果待测物质在紫外及可见光区没有明显吸收,而杂质有吸收,那么根据吸收曲线可检查出杂质。 根据摩尔吸光系数也可检查有无杂质。若待测物质有较强的吸收而所含杂质在此波长下基本无吸收,杂质的存在将使待测物质的摩尔吸光系数降低,反之亦然。

124
三、定量分析 依据:朗伯-比耳定律 吸光度: A= b c 透光度:-lgT = b c 灵敏度高:
max:104~105 L· mol-1 · cm -1;(比红外大) 测量误差与吸光度读数有关: A=0.434,读数相对误差最小;
 测量误差与吸光度读数有关: A=0.434,读数相对误差最小;")
125
标准曲线法 配制一系列标准品溶液 在待测物的max下,以适当的空白溶液作参比,测定各溶液的吸光度A。 以A为纵坐标,以浓度c为横坐标,绘制标准曲线,或由实验数据求出直线回归方程及相关系数。 用同样方法配制待测试样的溶液,在相同条件下测定其吸光度AX。 然后从标准曲线上查出与吸光度AX相对应的试样溶液的浓度cX,或由直线回归方程计算cX。

126
2. 直接比较法 在相同条件下配制标准溶液和待测试样溶液,浓度宜相近,以减少误差。然后分别测定它们的吸光度。根据吸收定律: 因K、b相同,故由此二式可得: 本法简便,但误差较大。分析时使cs与cx尽可能的接近,以提高测定结果的准确性。

127
3.双波长分光光度法 双波长分光光度法中,使交替照射到吸收池的两束波长分别为λ1和λ2的单色光的强度相等。待测组分对λ1和λ2的吸光度分别为A1和A2,背景吸收与光散射为AS,则 ΔA与溶液中待测组分的浓度c成正比。 只要λ1和λ2选择适当,就能将干扰组分的吸收消除掉,而不必预先分离或采用掩蔽手段。

128
用双波长分光光度法对两个组分混合物中某个组分的测定,常采用等吸收点法消除干扰。
等吸收点法是指在干扰组分的吸收光谱上,选两个适当的波长λ1和λ2, 干扰组分在这两个波长处具有相等的吸光度。

129
选择波长组合λ1 、λ2的基本要求是: ⑴选定的波长λ1和λ2处干扰组分应具有相同吸光度,即: ΔAy = ΔA yλ2 - ΔA yλ1 = 0 故: ΔAx+y = ΔA x=(εxλ2-εxλ1)bcx 此时:测得的吸光度差ΔA只与待测组分x的浓度呈线性关系,而与干扰组分y无关。若x为干扰组分,则也可用同样的方法测定y组分。 ⑵在选定的两个波长λ1和λ2处待测组分的吸光度应具有足够大的差值。 可采用作图法选择符合上述两个条件的波长组合。

130
4.导数光谱法 (derivative spectroscopy)
导数光谱法是解决干扰物质与待测物质的吸收光谱互相重叠,消除胶体和悬浮物散射影响和背景吸收的良好定量方法。 基本原理:根据吸收光谱的数据,每隔一个波长小间隔Δλ(一般为1~2nm)逐点计算出 的值。 绘制 ~λ曲线,得到的图形为一阶导数光谱。依次可求出高一阶的导数光谱,利用导数光谱对组分进行定性、定量分析 。 此法在多组分的同时测定、浑浊溶液测定、背景干扰消除及复杂光谱的辨析方面具有优越性。
逐点计算出 的值。 绘制 ~λ曲线,得到的图形为一阶导数光谱。依次可求出高一阶的导数光谱,利用导数光谱对组分进行定性、定量分析 。 此法在多组分的同时测定、浑浊溶液测定、背景干扰消除及复杂光谱的辨析方面具有优越性。")
131
导数光谱法的定量依据,根据Lambert-Beer定律得:
在一定条件下,吸光度A的n阶导数值与待测组分的浓度c成正比。 导数光谱法定量数据的测量:从导数光谱上选择适宜的导数信号,通常采用几何法测量出定量所需的数据。常用的测量方法有峰-谷法、基线法和峰-零法。

132
dI/dλ =-I0 bc dε/dλ 一阶导数信号与试样浓度呈线性关系; 测定灵敏度依赖于摩尔吸光系数对波长的变化率dε/dλ。吸收曲线的拐点处dε/dλ最大,故其灵敏度最高(见图)。 同理可以导出其二阶和三阶导数光谱(略)
。 同理可以导出其二阶和三阶导数光谱(略)")
133
四、有机化合物结构辅助解析 structure determination of organic compounds
1. 可获得的结构信息 (1) nm 无吸收峰。饱和化合物,单烯。 (2) nm有吸收峰(ε=10-100)醛酮 n→π* 跃迁产生的R 带。 (3) nm 有中等强度的吸收峰(ε= ),芳环的特征 吸收(具有精细解构的B带)。 (4) nm有强吸收峰(ε104),表明含有一个共轭体系(K)带。共轭二烯:K带(230 nm);不饱和醛酮:K带230 nm ,R带 nm 260nm,300 nm,330 nm有强吸收峰,3,4,5个双键的共轭体系。 ⑸若该有机物的吸收峰延伸至可见光区,则该有机物可能是长链共轭或稠环化合物。
 nm 无吸收峰。饱和化合物,单烯。 (2) nm有吸收峰(ε=10-100)醛酮 n→π* 跃迁产生的R 带。 (3) nm 有中等强度的吸收峰(ε= ),芳环的特征 吸收(具有精细解构的B带)。 (4) nm有强吸收峰(ε104),表明含有一个共轭体系(K)带。共轭二烯:K带(230 nm);不饱和醛酮:K带230 nm ,R带 nm. 260nm,300 nm,330 nm有强吸收峰,3,4,5个双键的共轭体系。 ⑸若该有机物的吸收峰延伸至可见光区,则该有机物可能是长链共轭或稠环化合物。")
134
2.光谱解析注意事项 (1) 确认max,并算出㏒ε,初步估计属于何种吸收带; (2) 观察主要吸收带的范围,判断属于何种共轭体系;
(3) 乙酰化位移 B带: 262 nm(ε302) nm(ε2040) nm(ε300) (4) pH值的影响 加NaOH红移→酚类化合物,烯醇。 加HCl兰移→苯胺类化合物。
 乙酰化位移. B带: 262 nm(ε302) 274 nm(ε2040) 261 nm(ε300) (4) pH值的影响. 加NaOH红移→酚类化合物,烯醇。 加HCl兰移→苯胺类化合物。")
135
3. 分子不饱和度的计算 定义: 不饱和度是指分子结构中达到饱和所缺一价元素的“对”数。 如:乙烯变成饱和烷烃需要两个氢原子,不饱和度为1。
计算: 若分子中仅含一,二,三,四价元素(H,O,N,C),则可按下式进行不饱和度的计算: = (2 + 2n4 + n3 – n1 )/ 2 n4 , n3 , n1 分别为分子中四价,三价,一价元素数目。 作用: 由分子的不饱和度可以推断分子中含有双键,三键,环,芳环的数目,验证谱图解析的正确性。 例: C9H8O2 = (2 +29 – 8 )/ 2 = 6
,则可按下式进行不饱和度的计算: = (2 + 2n4 + n3 – n1 )/ 2. n4 , n3 , n1 分别为分子中四价,三价,一价元素数目。 作用: 由分子的不饱和度可以推断分子中含有双键,三键,环,芳环的数目,验证谱图解析的正确性。 例: C9H8O2. = (2 +29 – 8 )/ 2 = 6.")
136
4. 解析示例 有一化合物C10H16由红外光谱证明有双键和异丙基存在,其紫外光谱 max=231 nm(ε 9000),此化合物加氢只能吸收2克分子H2,,确定其结构。 解:①计算不饱和度 = 3;两个双键;共轭?加一分子氢 ②max=231 nm, ③可能的结构 ④计算 max max: max =非稠环二烯(a,b)+2 × 烷基取代+环外双键 =217+2×5+5=232(231)
+2 × 烷基取代+环外双键. =217+2×5+5=232(231)")
137
吸收波长计算

138
立体结构和互变结构的确定 顺式:λmax=280nm; εmax=10500 反式:λmax=295.5 nm;εmax=29000
互变异构: 酮式:λmax=204 nm;无共轭 烯醇式:λmax=243 nm

139
取代苯吸收波长计算

140
§8 催化动力学分光光度法 催化动力学分光光度法(spectrophotometry by catalytic kinetics)是以催化反应为基础,通过测定催化体系中反应物或产物的吸光度A(A间接反映反应速度)来确定催化剂含量的方法。该法的灵敏度高、检测限低,方法简便,易于推广,广泛应用于生物样品及环境样品中痕量元素的测定。
是以催化反应为基础,通过测定催化体系中反应物或产物的吸光度A(A间接反映反应速度)来确定催化剂含量的方法。该法的灵敏度高、检测限低,方法简便,易于推广,广泛应用于生物样品及环境样品中痕量元素的测定。")
141
一、指示反应与指示物质 化学反应速率依赖于催化剂(或抑制剂)的浓度,若利用此反应可以测定催化剂(或抑制剂)的量,这种化学反应称为指示反应。指示反应中的反应物和生成物称为指示物质。 如碘离子催化Ce(Ⅳ)氧化As(Ⅲ)生成Ce(Ⅲ)和As(Ⅴ)的反应,此反应可测定催化剂碘离子的量,该反应称为碘离子的指示反应,Ce(Ⅳ)、As(Ⅲ)、Ce(Ⅲ)和As(Ⅴ)均为指示物质。 最常用的指示反应是氧化还原反应。常用的氧化剂为H2O2、Ce(Ⅳ)、卤酸盐,还原剂多为有机化合物,催化剂为金属离子。
氧化As(Ⅲ)生成Ce(Ⅲ)和As(Ⅴ)的反应,此反应可测定催化剂碘离子的量,该反应称为碘离子的指示反应,Ce(Ⅳ)、As(Ⅲ)、Ce(Ⅲ)和As(Ⅴ)均为指示物质。 最常用的指示反应是氧化还原反应。常用的氧化剂为H2O2、Ce(Ⅳ)、卤酸盐,还原剂多为有机化合物,催化剂为金属离子。")
142
设一显色反应的反应速率较慢,在催化剂的催化下速度加快,其催化显色反应为:
二、定量依据 设一显色反应的反应速率较慢,在催化剂的催化下速度加快,其催化显色反应为: 若选择产物F为指示物质,根据质量作用定律,速率方程为 控制实验条件,使cA、cB的量很大,其浓度的改变可以忽略不计 ,即cA、cB为常数,与Ka合并为Kb,则为 aA + bB fF +gG 催化剂

143
积分后得: 产物F的吸光度与其浓度c催符合Lambert-Beer定律,即 在实验条件一定时,ε、b为常数,与Kb合并得 定量依据

144
三、定量方法 在具体测定时,常采用固定时间法,此时t也为常数,t与K‘合并为K,则 A与c催成线性关系。催化动力学分光光度法多采用快速冷却终止指示反应,标准溶液及待测溶液均在同样的时间内进行,通过工作曲线法求出待测物质c催的量。

145
一、 示差分光光度法 §9 提高分析灵敏度和准确度的方法 1. 高浓度示差法
§9 提高分析灵敏度和准确度的方法 一、 示差分光光度法 1. 高浓度示差法 设:待测溶液浓度为cx,标准溶液浓度为cs(cs < cx)。 则: Ax= Kb cx As = Kb cs Ar = ΔA =Ax -As = Kb(cx - cs ) ΔA = KbΔc 绘制ΔA ~Δc工作曲线,根据cx=cs+Δc计算试样浓度
。 则: Ax= Kb cx. As = Kb cs. Ar = ΔA =Ax -As = Kb(cx - cs ) ΔA = KbΔc. 绘制ΔA ~Δc工作曲线,根据cx=cs+Δc计算试样浓度.")
146
示差法为什么能较准确地测定高浓度组分呢?
普通光度法: cs: Ts = 10%, cx : Tx=5%, 示差法: cs : Ts =100%, cx : Tr=50%, 透射比读数标尺扩大了十倍。读数落入适宜读数范围内,提高了测量的准确度。

147
示差法的浓度相对误差: 随着参比溶液浓度增加,即Ts 减小,浓度相对误差减小。
示差法要求仪器光源有足够的发射强度或能增大光电流放大倍数,以便能调节参比溶液透射比为 100%。

148
2. 低浓度示差法 适用:A<0.1 方法:选用浓度稍高于待测溶液的标准溶液校正仪器,调节透光度为0,用空白溶液调节透光度为100%,然后测量待测溶液的吸光度。

149
二、萃取分光光度法 溶剂萃取与分光光度法相结合的方法。 优点:具有富集作用,可消除干扰离子,提高方法的灵敏度和选择性。

150
三、胶束增溶分光光度法 1. 三元配合物 稳定,可提高分析测定的准确度和重现性 比二元配合物具有更高的灵敏度和更大的对比度
1. 三元配合物 由一种金属离子同时与两种不同的配位体形成的三元配合物具有下列分析特性: 稳定,可提高分析测定的准确度和重现性 例: Ti-EDTA-H2O2三元配合物的稳定性,比Ti-EDTA和Ti-H2O2二元配合物的稳定性,分别增强约1000倍和100倍。 比二元配合物具有更高的灵敏度和更大的对比度 灵敏度通常可提高1~2倍,有时甚至提高5倍以上。 比二元体系具有更高的选择性 减少了金属离子形成类似配合物的可能性。

151
2.胶束增溶分光光度法 在二元配合物中加入某些表面活性剂可与金属离子及显色剂形成三元胶束状配合物。 胶束:疏水基团在水溶液中相互聚集成为几十个分子组成的胶体质点。 胶束的作用:增溶;增稳;增敏 常用表面活性剂:氯化十六烷基三甲胺、溴化十六烷基三甲胺、溴化十六烷基吡啶、溴化十四烷基吡啶

152
§10 分光光度法的应用 一、样品中组分的定量测定 见教材P209~210

153
二、 光度滴定 光度测量用来确定滴定终点灵敏,并可克服目视滴定法中的干扰。 终点由直线外推法得到,可用于平衡常数较小的滴定反应。

154
三、 酸碱离解常数的测定 用于测定对光有吸收的弱酸(碱)的离解常数。 HL = H++L-
(1) 配制一系列总浓度(c)相等,而pH不同的HL溶液 (2) 测定各溶液的pH值。 (3)在酸式(HL)或碱式(L-)最大吸收波长处,测吸光度。
 配制一系列总浓度(c)相等,而pH不同的HL溶液. (2) 测定各溶液的pH值。 (3)在酸式(HL)或碱式(L-)最大吸收波长处,测吸光度。")
155
(4)假设高酸度时,弱酸全部以酸式形式存在,则: (5)在低酸度时,弱酸全部以碱式形式存在,则: AL= κLc
根据分布系数的概念 (4)假设高酸度时,弱酸全部以酸式形式存在,则: AHL= κHLc (5)在低酸度时,弱酸全部以碱式形式存在,则: AL= κLc
假设高酸度时,弱酸全部以酸式形式存在,则: AHL= κHLc. (5)在低酸度时,弱酸全部以碱式形式存在,则: AL= κLc.")
156
光度法测定一元弱酸离解常数的基本公式。

157
四、 配合物组成及稳定常数的测定 摩尔比法 M + nL = MLn
(1) 测定金属离子浓度 cM固定,而配位体浓度cL逐渐改变的不同溶液的吸光度。作图 (2)当cL/cM<n时,配位不完全 当cL/cM>n时,配位完全
 测定金属离子浓度 cM固定,而配位体浓度cL逐渐改变的不同溶液的吸光度。作图. (2)当cL/cM<n时,配位不完全. 当cL/cM>n时,配位完全.")
158
思考题 1. 为什么物质对光会发生选择性吸收? 2. 朗伯-比耳定律的物理意义是什么?什么是透射比?什么是吸光度?二者之间的关系是什么?
3. 摩尔吸收系数的物理意义是什么?其大小和哪些因素有关?在分析化学中有何意义? 4. 什么是吸收光谱曲线?什么是标准曲线?它们有何实际意义?利用标准曲线进行定量分析时可否使用透射比T和浓度c为坐标?

159
思考题 5. 当研究一种新的显色剂时,必须作哪些实验条件的研究?为什么? 6. 分光光度计有哪些主要部件?它们各起什么作用?
7. 测定金属钴中微量锰时,在酸性液中用KIO3将锰氧化为高锰酸根离子后进行吸光度的测定。若用高锰酸钾配制标准系列,在测定标准系列及试液的吸光度时应选什么作参比溶液?

160
思考题 8. 吸光度的测量条件如何选择?为什么?普通光度法与示差法有何异同?
9. 光度分析法误差的主要来源有哪些?如何减免这些误差?试根据误差分类分别加以讨论。 10. 常见的电子跃迁有哪几种类型? 11. 在有机化合物的鉴定和结构判断上,紫外-可见吸收光谱提供信息具有什么特点?












>")

>")

 紫外分光光度法测定食盐中的碘含量.>")





 清楚水资源及其水质污染监测的对象、目的、监测项目和主要的水质监测分析方法; 2) 掌握水质在物理性质、金属化合物和非金属无机物、有机化合物监测的项目及监测方法; 3) 掌握水质监测方案制订、水样 采集保存和预处理以及底质监测。>")
