Download presentation
Presentation is loading. Please wait.

1
5 分子生物学研究法(上) 从20世纪中叶开始,分子生物学研究得到高速发展,除了基础理论的重大突破,主要原因之一是研究方法,特别是基因操作和基因工程技术的进步。
 从20世纪中叶开始,分子生物学研究得到高速发展,除了基础理论的重大突破,主要原因之一是研究方法,特别是基因操作和基因工程技术的进步。")
2
基因操作主要包括DNA分子的切割与连接、核酸分子杂交、凝胶电泳、细胞转化、核酸序列分析以及基因人工合成、定点突变和PCR扩增等,是分子生物学研究的核心与基本技术。

3
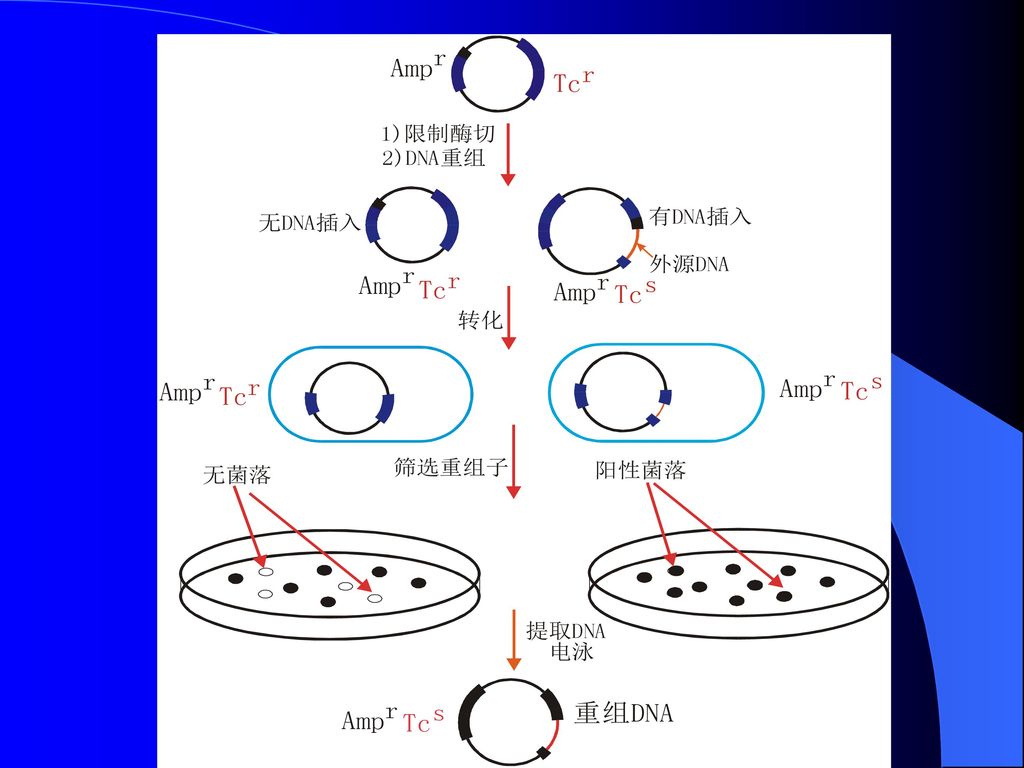
基因工程是指在体外将核酸分子插入病毒、质粒或其它载体分子,构成遗传物质的新组合,使之进入原先没有这类分子的寄主细胞内并进行持续稳定的繁殖和表达。

4
基因工程技术是核酸操作技术的一部分,只不过它强调了外源核酸分子在另一种不同的寄主细胞中的繁衍与性状表达。
事实上,这种跨越物种屏障、把来自其它生物的基因置于新的寄主生物细胞之中的能力,是基因工程技术区别于其它技术的根本特征。

6
5. 1 重组DNA技术回顾 三大成就: 第一,在20世纪40年代确定了遗传信息的携带者、即基因的分子载体是DNA而不是蛋白质——解决了遗传的物质基础问题; 第二,50年代提出了DNA分子的双螺旋结构模型和半保留复制机制——解决了基因的自我复制和世代交替问题;

7
第三,50年代末至60年代,相继提出了“中心法则”和操纵子学说,成功地破译了遗传密码——阐明了遗传信息的流动与表达机制。

8
表5-1 重组DNA技术史上的主要事件 年 份 事 件 1869 F Miescher首次从莱茵河鲑鱼精子中分离DNA。 1957
年 份 事 件 1869 F Miescher首次从莱茵河鲑鱼精子中分离DNA。 1957 A.Kornberg从大肠杆菌中发现了DNA聚合酶I。 Ochoa发现RNA聚合酶和信使RNA,并证明mRNA决定了蛋白质分子中的氨基酸序列。 1961 Nirenberg破译了第一个遗传密码;Jacob和Monod提出了调节基因表达的操纵子模型。 1964 Yanofsky和Brenner等人证明,多肽链上的氨基酸序列与该基因中的核苷酸序列存在着共线性关系。 1965 Holley完成了酵母丙氨酸tRNA的全序列测定;科学家证明细菌的抗药性通常由“质粒”DNA所决定。

9
1966 Nirenberg,Ochoa,Khorana,Crick等人破译了全部遗传密码。 1967 第一次发现DNA连接酶 1970 Smith,Wilcox和Kelley分离了第一种限制性核酸内切酶,Temin和Baltimore从RNA肿瘤病毒中发现反转录酶。 Boyer,Berg等人发展了DNA重组技术,于72年获得第一个重组DNA分子,73年完成第一例细菌基因克隆。 Sanger与Maxam和Gilbert等人发明了DNA序列测定技术,1977年完成了全长5387bp的噬菌体φx174基因组测定。 1978 首次在大肠杆菌中生产由人工合成基因表达的人脑激素和人胰岛素。 1980 美国联邦最高法院裁定微生物基因工程可以被专利化。 1981 Palmiter和Brinster获得转基因小鼠,Spradling和Rubin得到转基因果蝇。 1982 美、英批准使用第一例基因工程药物——胰岛素,Sanger等人完成了入噬菌体48,502bp全序列测定。

10
1983 获得第一例转基因植物。 1984 斯坦福大学获得关于重组DNA的专利。 1986 GMO首次在环境中释放。 1988 Watson出任“人类基因组计划”首席科学家。 1989 DuPont公司获得转肿瘤基因小鼠—“Oncomouse”。 1992 欧共体35个实验室联合完成酵母第三染色体全序列测定(315kb)。 1994 第一批基因工程西红柿在美国上市。 1996 完成了酵母基因组(1.25×107bp)全序列测定。 1997 英国爱丁堡罗斯林研究所获得克隆羊。 2000 完成第一个高等植物拟南芥的全序列测定((1.15×108bp)。 2001 完成第一个人类基因组全序列测定(2.7×109bp)。
。 第一批基因工程西红柿在美国上市。 完成了酵母基因组(1.25×107bp)全序列测定。 英国爱丁堡罗斯林研究所获得克隆羊。 完成第一个高等植物拟南芥的全序列测定((1.15×108bp)。 完成第一个人类基因组全序列测定(2.7×109bp)。")
11
(1)限制性核酸内切酶 限制性核酸内切酶能够识别DNA上的特定碱基序列并从这个位点切开DNA分子。
第一个核酸内切酶Eco RI是Boyer实验室在1972年发现的,它能特异性识别GAATTC序列,将双链DNA分子在这个位点切开并产生具有粘性末端或平端的小片段。

12
图5-1 几种主要DNA内切酶所识别的序列及其酶切末端。

13
(2)DNA连接酶 图5-2 DNA连接酶能把不同的DNA片段连接成一个整体。a. DNA的粘性末端; b. DNA的平末端; c. 化学合成的具有EcoRI粘性末端的DNA片段。
DNA连接酶 图5-2 DNA连接酶能把不同的DNA片段连接成一个整体。a. DNA的粘性末端; b. DNA的平末端; c. 化学合成的具有EcoRI粘性末端的DNA片段。")
14
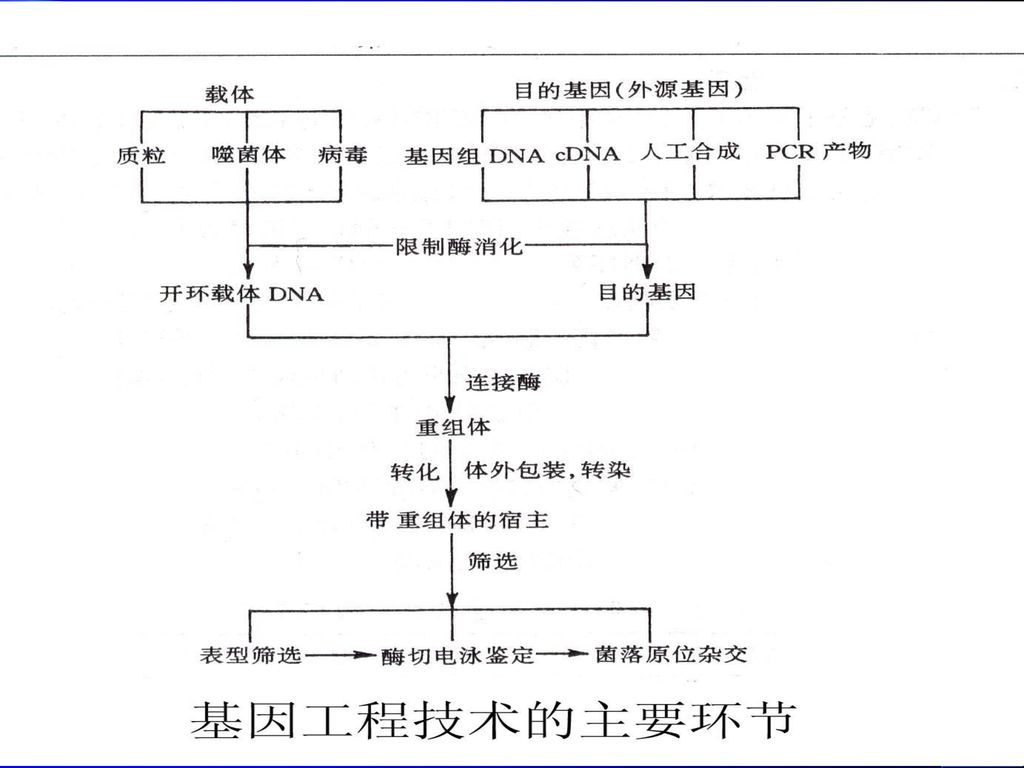
(3)分子克隆的载体 仅仅能在体外利用限制性核酸内切酶和DNA连接酶进行DNA的切割和重组,还不能满足基因工程的要求,只有将它们连接到具备自主复制能力的DNA分子上,才能在寄主细胞中进行繁殖。 具备自主复制能力的DNA分子就是分子克隆的载体(vector)。病毒、噬菌体和质粒等小分子量复制子都可以作为基因导入的载体。
。病毒、噬菌体和质粒等小分子量复制子都可以作为基因导入的载体。")
15
图5-3 重组DNA操作过程示意图

16
目的基因的表达

21
5. 2 DNA基本操作技术 核酸凝胶电泳技术 自从琼脂糖(agarose)和聚丙烯酰胺(polyacrylamide)凝胶被引入核酸研究以来,按分子量大小分离DNA的凝胶电泳技术,已经发展成为一种分析鉴定重组DNA分子及蛋白质与核酸相互作用的重要实验手段。
和聚丙烯酰胺(polyacrylamide)凝胶被引入核酸研究以来,按分子量大小分离DNA的凝胶电泳技术,已经发展成为一种分析鉴定重组DNA分子及蛋白质与核酸相互作用的重要实验手段。 .")
22
一种分子被放置到电场中,它就会以一定的速度移向适当的电极。我们把这种电泳分子在电场作用下的迁移速度,叫做电泳的迁移率,它与电场强度和电泳分子本身所携带的净电荷数成正比,与片段大小成反比。

23
生理条件下,核酸分子中的磷酸基团呈离子化状态,所以,DNA和RNA又被称为多聚阴离子(polyanions),在电场中向正电极的方向迁移。
,在电场中向正电极的方向迁移。")
24
琼脂糖凝胶分辨DNA片段的范围为0.2~50kb之间。
聚丙烯酰胺凝胶的分辨范围为1到1000个碱基对之间。 凝胶浓度的高低影响凝胶介质孔隙的大小,浓度越高,孔隙越小,其分辨能力就越强。

25
表5-2 琼脂糖及聚丙烯酰胺凝胶分辨DNA片段的能力
凝胶类型及浓度 分离DNA的大小范围(bp) 0.3%琼脂糖 ~1 000 0.7%琼脂糖 ~1 000 1.4%琼脂糖 ~300 4.0%聚丙烯酰胺 ~100 10.0%聚丙烯酰胺 ~25 20.0%聚丙烯酰胺 ~1
 0.3%琼脂糖 ~ %琼脂糖 ~ %琼脂糖 6 000~ %聚丙烯酰胺 1 000~ %聚丙烯酰胺 500~ %聚丙烯酰胺 50~1.")
26
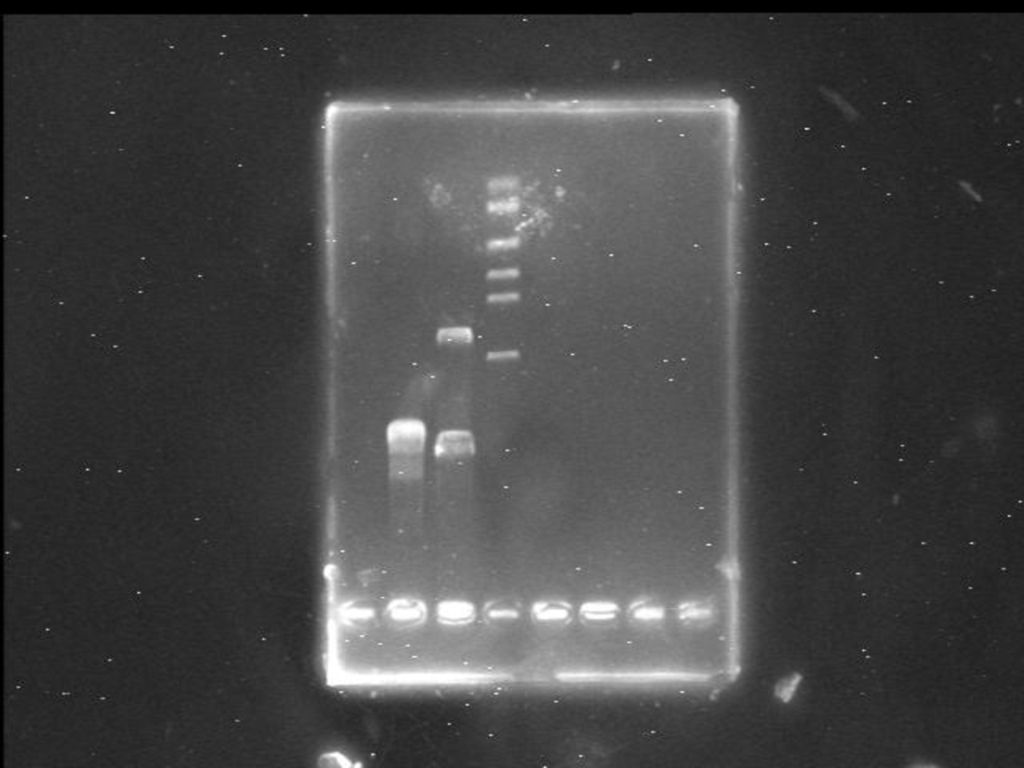
在凝胶电泳中,加入溴化乙锭(ethidium bromide,EtBr)染料对核酸分子进行染色,然后放置在紫外光下观察,可灵敏而快捷地检测出凝胶介质中DNA的谱带部位,即使每条DNA带中仅含有0.05 g的微量DNA,也可以被清晰地检测出来。
染料对核酸分子进行染色,然后放置在紫外光下观察,可灵敏而快捷地检测出凝胶介质中DNA的谱带部位,即使每条DNA带中仅含有0.05 g的微量DNA,也可以被清晰地检测出来。")
27
溴化乙锭是一种具扁平分子的核酸染料,能插入到DNA或RNA分子的相邻碱基之间,并在300 nm波长的紫外光照射下发出荧光。

28
图5-4 溴化乙锭染料的化学结构及其对DNA分子的插入作用。 由于插入了溴化乙锭分子,在紫外光照射下,琼脂糖凝胶

30
5.2.2 细菌转化与目标DNA分子的增殖 所谓细菌转化,是指一种细菌菌株由于捕获了来自另一种细菌菌株的DNA而导致性状特征发生遗传改变的生命过程。 提供转化DNA的菌株叫作供体菌株,接受转化DNA的细菌菌株则被称为受体菌株。

31
转化(transformation) 特指以质粒DNA或以它为载体构建的重 组子导入细菌的过程。 转染 (transfection) 是指噬菌体、病毒或以它作为载体构成的重组子导入细胞的过程。 转导 (transduction) 以噬菌体为媒介,将外源DNA导入细菌的过程。 上述概念往往容易混淆。
 以噬菌体为媒介,将外源DNA导入细菌的过程。 上述概念往往容易混淆。")
32
转化是一个自然存在的过程。细菌处于容易吸收外源DNA状态叫感受态,用理化方法诱导细胞进入感受态的操作叫致敏过程。
重组DNA转化细菌的技术操作关键就是通过化学方法,人工诱导细菌细胞进入一个敏感的感受态,以便外源DNA进入细菌内。 这项技术始于Mandel和Higa 1970年的观察,他们发现细菌经过冰冷的CaCl2溶液处理及短暂热休克后,容易被λ噬菌体DNA感染,随后Cohn于 1972年进一步证明质粒DNA用同样的方法也可进入细菌。

33
转化原理: 将快速生长中的大肠杆菌置于经0℃预处理的低渗氯化钙溶液中,使细胞膨胀,细胞膜通透性发生变化,易与外源DNA相粘附并在细胞表面的复合物。 42℃下做短暂热刺激,复合物便会被细胞所吸收。在全培养基中生长一段时间使转化基因实现表达,就可涂布于选择性培养基中分离转化子。 细菌转化及蓝白斑筛选:图5-5

34
图5-6 λ噬菌体可以作为克隆载体

36
聚合酶链反应技术(PCR) 首先将双链DNA分子加热分离成两条单链,DNA聚合酶以单链DNA为模板并利用反应混合物中的四种dNTP合成新生的DNA互补链。 因为DNA聚合酶需要有一小段双链DNA来启动(“引导”)新链的合成,所以,新生DNA链的起点由寡核苷酸引物在模板DNA链两端的退火位点所决定。
新链的合成,所以,新生DNA链的起点由寡核苷酸引物在模板DNA链两端的退火位点所决定。")
37
由于在PCR反应中所选用的一对引物,是按照与扩增区段两端序列彼此互补的原则设计的,因此每一条新生链的合成都是从引物的退火结合位点开始并朝相反方向延伸的。

38
整个PCR反应的全过程,即DNA解链(变性)、引物与模板DNA相结合(退火)、DNA合成(链的延伸)三步,可以被不断重复。经多次循环之后,反应混合物中所含有的双链DNA分子数,即两条引物结合位点之间的DNA区段的拷贝数,理论上的最高值应是2n。
、引物与模板DNA相结合(退火)、DNA合成(链的延伸)三步,可以被不断重复。经多次循环之后,反应混合物中所含有的双链DNA分子数,即两条引物结合位点之间的DNA区段的拷贝数,理论上的最高值应是2n。")
39
图5-8 PCR指数扩增时循环次数与DNA产物数量的比较。

40
主要步骤: 将含有待扩增DNA样品的反应混合物放置在高温(>94℃)环境下加热1分钟,使双链DNA变性,形成单链模板DNA。
环境下加热1分钟,使双链DNA变性,形成单链模板DNA。")
41
然后降低反应温度(约50℃),致冷1分钟,使寡核苷酸引物与两条单链模板DNA发生退火作用并结合在靶DNA区段两端的互补序列位置上。
,致冷1分钟,使寡核苷酸引物与两条单链模板DNA发生退火作用并结合在靶DNA区段两端的互补序列位置上。")
42
最后,将反应混合物的温度上升到72℃左右保温1-数分钟,在DNA聚合酶的作用下,从引物的3'-端加入脱氧核苷三磷酸,并沿着模板分子按5'→3'方向延伸,合成新生DNA互补链。

43
聚合酶链式反应示意图。(a)起始材料,双链DNA;(b)反应混合物加热后发生链分离,降温使引物结合到待扩增靶DNA区段两端的退火位点上;(c)Taq聚合酶以单链DNA为模板在引物的引导下利用反应混合物中的dNTPs合成互补的新链DNA;(d)将反应混合物再次加热,使旧链和新链分开;(e)合成新的互补链DNA;(f)重复b;(g)重复c、、、、、、
起始材料,双链DNA;(b)反应混合物加热后发生链分离,降温使引物结合到待扩增靶DNA区段两端的退火位点上;(c)Taq聚合酶以单链DNA为模板在引物的引导下利用反应混合物中的dNTPs合成互补的新链DNA;(d)将反应混合物再次加热,使旧链和新链分开;(e)合成新的互补链DNA;(f)重复b;(g)重复c、、、、、、")
44
引物设计一般遵循下列原则: (1)引物长度以15~30 bp为宜。
(2)引物碱基尽可能随机分布,避免出现嘌呤、嘧啶堆积现象,引物G+C含量宜在45~55%左右。 (3)引物内部不应形成二级结构,两个引物之间尤其在3'末端不应有互补链存在。 (4)引物的碱基顺序不应与非扩增区域有同源性。要求在引物设计时采用计算机进行辅助检索分析。 (5)引物3‘末端碱基:原则上要求引物3’末端与模板DNA一定要配对。 (6)引物5'末端碱基:PCR反应物5'末端碱基并没有严格的限制,只要与模板DNA结合的引物长度足够,其5'末端碱基可以不与模板DNA匹配而呈游离状态。
引物碱基尽可能随机分布,避免出现嘌呤、嘧啶堆积现象,引物G+C含量宜在45~55%左右。 (3)引物内部不应形成二级结构,两个引物之间尤其在3 末端不应有互补链存在。 (4)引物的碱基顺序不应与非扩增区域有同源性。要求在引物设计时采用计算机进行辅助检索分析。 (5)引物3‘末端碱基:原则上要求引物3’末端与模板DNA一定要配对。 (6)引物5 末端碱基:PCR反应物5 末端碱基并没有严格的限制,只要与模板DNA结合的引物长度足够,其5 末端碱基可以不与模板DNA匹配而呈游离状态。")
45
PCR反应引物设计: PCR作为一个体外酶促反应,其效率和特异性取决于两个方面: 一是引物与模板的特异结合 二是多聚酶对引物的有效延伸 基因组DNA作为模板时,由于其数量的庞大及结构复杂,除了特异扩增外,往往很容易产生非特异性扩增产物。引物设计的总原则就是提高扩增的效率和特异性。

46
5. 2. 4 实时定量PCF(Real time-PCR)
实时定量PCR技术:利用带荧光检测的PCR仪对PCR过程DNA累积作出动态监测。 荧光染料: SYBR Green I:图5-9 TaqMan探针:图5-10

47
5. 2. 5 基因组DNA文库构建 基因组 DNA文库构建:把某种生物基因组DNA切成适当大小,分别与载体组合,导入微生物细胞,形成克隆。
常用载体:噬菌体;柯斯质粒;BAC;PAC;YAC

48
5. 3 RNA基本操作技术 在DNA水平上分离真核生物的靶基因难度大:基因组DNA庞大;大量重复序列;有内含子。
而分离mRNAcDNA目的基因工作简单,速度快。 但是,RNA分子:敏感,稳定性差,某些基因的mRNA具有时相和转录的特殊性。

49
5. 3.1 总RNA的提取 1、操作要求高:选择合适的时相、采用适当的条件与材料,避免降解。 2、方法:
Trizol法。由苯酚和异硫氰酸胍组成,可迅速破坏细胞结构,使RNA释放,并保持RNA完整。 3、浓度和纯度鉴定: 通过OD260/OD280的比值来鉴定

50
5. 3.2 mRNA的纯化 根据mRNA3’端具有polyA尾巴的特点,利用寡聚(dT)-纤维素柱色谱法纯化获得高纯度的mRNA。
图5-14

51
图5-14 PolyATtract mRNA的分离纯化过程简图

52
5.3.3 cDNA的合成 使用oligo dT或随机引物,通过RT-PCR法(——逆转录酶)。 图5-15
。 图5-15")
53
图5-15 cDNA合成过程示意图

54
5.3.4 cDNA文库的构建 获得的含有某种组织器官cDNA信息的噬菌体文库,可用于筛选目的基因、大规模测序、基因芯片杂交等功能。
做法:mRNAcDNA 载体大肠杆菌 一个完整的cDNA文库通常包含大于500000的独立克隆。

55
5.3.5 基因文库的筛选 通过某种特定的方法从基因文库中鉴定出含有所需DNA分子的特定克隆过程。
1、核酸杂交法(Sourthern blotting):图5-17 2、PCR筛选法 3、免疫筛选法(Western blotting):该法适用于对表达文库的筛选。图5-18
:图 、PCR筛选法. 3、免疫筛选法(Western blotting):该法适用于对表达文库的筛选。图5-18.")
56
5.4 SNP的理论与应用 SNP:single nucleotide polymorphism,单核苷酸多态性。
具有很高的遗传稳定性,是继限制性片段长度多态性(RFLP)、RAPD和微卫星DNA(SSR)标记之后的第三代遗传标记。
、RAPD和微卫星DNA(SSR)标记之后的第三代遗传标记。")
57
RFLP(Restriction Fragment Length Polymorphisma)技术
RFLP标记是发展最早的DNA标记技术。RFLP是指基因型之间限制性片段长度的差异,这种差异是由限制性酶切位点上碱基的插入、缺失、失重排或点突变所引起的。 RFLP技术主要包括以下基本步骤: DNA提取→用限制性内切酶酶切DNA→用凝胶电泳分开DNA片段→把DNA片段转移到滤膜上→利用放射性标记的探针杂交显示特定的DNA片段(Southern杂交)和结果分析。
和结果分析。")
60
RAPD技术 RAPD((Random Amplified Polymorphic DNA)技术是建立在PCR (Polymerase Chain Reaction)基础之上的一种可对整个未知序列的基因组进行多态性分析的分子技术。其以基因组DNA为模板, 以单个人工合成的随机多态核苷酸序列( 通常为10 个碱基对) 为引物, 在热稳定的DNA 聚合酶( Taq 酶) 作用下, 进行PCR 扩增。扩增产物经琼脂糖或聚丙烯酰胺电泳分离、溴化乙锭染色后,在紫外透视仪上检测多态性。扩增产物的多态性反映了基因组的多态性。RAPD 技术现已广泛的应用于生物的品种鉴定、系谱分析及进化关系的研究上 。
技术是建立在PCR (Polymerase Chain Reaction)基础之上的一种可对整个未知序列的基因组进行多态性分析的分子技术。其以基因组DNA为模板, 以单个人工合成的随机多态核苷酸序列( 通常为10 个碱基对) 为引物, 在热稳定的DNA 聚合酶( Taq 酶) 作用下, 进行PCR 扩增。扩增产物经琼脂糖或聚丙烯酰胺电泳分离、溴化乙锭染色后,在紫外透视仪上检测多态性。扩增产物的多态性反映了基因组的多态性。RAPD 技术现已广泛的应用于生物的品种鉴定、系谱分析及进化关系的研究上 。")
62
SSR(simple sequence repeats)技术
微卫星DNA:重复单位序列最短,只有2~6bp,串联成簇,长度50~100bp,又称为短串联重复序列(Short Tandem Repeat STR)。广泛分布于基因组中。 SSR标记的基本原理: 根据微卫星序列两端互补序列设计引物,通过PCR反应扩增微卫星片段,由于核心序列串联重复数目不同,因而能够用PCR的方法扩增出不同长度的PCR产物,将扩增产物进行凝胶电泳,根据分离片段的大小决定基因型并计算等位基因频率。
。广泛分布于基因组中。 SSR标记的基本原理: 根据微卫星序列两端互补序列设计引物,通过PCR反应扩增微卫星片段,由于核心序列串联重复数目不同,因而能够用PCR的方法扩增出不同长度的PCR产物,将扩增产物进行凝胶电泳,根据分离片段的大小决定基因型并计算等位基因频率。")
63
SNP检测技术 传统SNP检测方法:RFLP、PCR-SSCP、DHPLC等,最终均需通过凝胶电泳分析,通量受到限制,并只能判断有无,而不知碱基类型。必须进行DNA测序。 基因分型:利用数据库已有SNP进行特定人群序列和发生频率研究,包括如下方法:

64
1、基因芯片技术:通过优化芯片杂交程序,使探针只与完全互补的序列杂交,而不与含有单个错配碱基序列杂交。
2、Taqman技术:运用了荧光共振能量转换(FRET)技术。 3、分子信标技术:也运用了FRET技术。 4、焦磷酸测序法
技术。 3、分子信标技术:也运用了FRET技术。 4、焦磷酸测序法.")
65
SNP的应用 1、人类基因单体的绘图 2、SNP与疾病易感基因的相关性分析 3、指导用药与药物设计

66
5.5 基因克隆技术 在多细胞的高等生物个体水平上,人们用克隆(clone)表示由具有相同基因型的同一物种的两个或数个个体组成的群体。
在细胞水平上,克隆是指由同一个祖细胞分裂而来的一群带有完全相同遗传物质的子细胞。

67
在分子生物学中,人们把将外源DNA插入具有复制能力的载体DNA中,使之得以永久保存和复制的过程称为克隆(动词)。
。")
68
基因克隆,包括目的基因的分离和鉴定两个内容,分四个基本步骤:
(1) DNA材料的选择与片段化; (2) 外源DNA片段与载体分子的体外连接反应; (3) 将人工重组的DNA分子导入它们能够进行正常复制的寄主细胞; (4) 重组转化子克隆的选择或筛选。 *真核生物基因(大,有重复序列)———cDNA克隆法法。
 DNA材料的选择与片段化; (2) 外源DNA片段与载体分子的体外连接反应; (3) 将人工重组的DNA分子导入它们能够进行正常复制的寄主细胞; (4) 重组转化子克隆的选择或筛选。 *真核生物基因(大,有重复序列)———cDNA克隆法法。")
69
RACE技术 RACE(Raid amplification of cDNA ends)技术是一项在已知cDNA序列(部分)的基础上克隆5’或3’端缺失序列的技术。 主要操作步骤如图5-22所示。 RACE技术除了用于获得全长cDNA序列外,还被应用于识别获得5’和3’端非转录序列。

70
应用cDNA差示分析法克隆基因 该法通过降低cDNA群体复杂性和更换cDNA两端接头等方法,特异性扩增目的基因片段。因为Tester和Driver在接受差示分析前均经一个4碱基切割酶处理,形成平均长度256bp的代表群,保证绝大部分遗传信息能被扩增。

71
图5-23 RDA流程图。

72
5.5.3 Gateway大规模克隆技术 1、基因大规模克隆的目的
Gateway技术利用噬菌体进行位点特异性DNA片段重组,实现不需要传统的酶切连接过程的基因快速克隆和载体间平行转移,为大规模克隆基因组注释的可译框架提供了保障。 2、Gateway克隆技术要点 (1)Topo反应:将目的基因PCR产物连入Entry载体 (2) LR反应:将目的片断从Entry载体重组入表达载体 图5-24a,5-24b。
Topo反应:将目的基因PCR产物连入Entry载体. (2) LR反应:将目的片断从Entry载体重组入表达载体. 图5-24a,5-24b。")
73
图5-24 Gateway大规模克隆策略

74
5.5.4 基因的图位克隆法 基因的图位克隆法(Map-based cloning)是分离未知性状目的基因的一种良好方法。
基因的图位克隆法 基因的图位克隆法(Map-based cloning)是分离未知性状目的基因的一种良好方法。 所有具有某种表现型的基因都可以通过该方法克隆得到。首先,通过构建遗传连锁图,将目的基因定位到某个染色体的特定位点,并在其两侧确定紧密连锁的RFLP或RAPD分子标记。
是分离未知性状目的基因的一种良好方法。 所有具有某种表现型的基因都可以通过该方法克隆得到。首先,通过构建遗传连锁图,将目的基因定位到某个染色体的特定位点,并在其两侧确定紧密连锁的RFLP或RAPD分子标记。")
75
其次,通过对许多不同的生态型及大量限制性内切酶和杂交探针的分析,找出与目的基因距离最近的RFLP标记,通过染色体步移技术将位于这两个标记之间的基因片段克隆并分离出来(图5-25)。
。")
76
图5-25 染色体步移法克隆基因示意图

77
图5-26 用图位克隆法获得控制水稻脆杆基因BCl

78
5.6 蛋白质组与蛋白质组学技术 1995年,首次提出了蛋白组学的概念。其是蛋白质和基因组研究在形式和内容方面的结合,致力于研究某一物种、个体、器官或细胞在特定条件、特定时间所表达的全部蛋白质图谱。 基因组是相对固定的,但各个基因的表达调控与表达程度却有时空性。

79
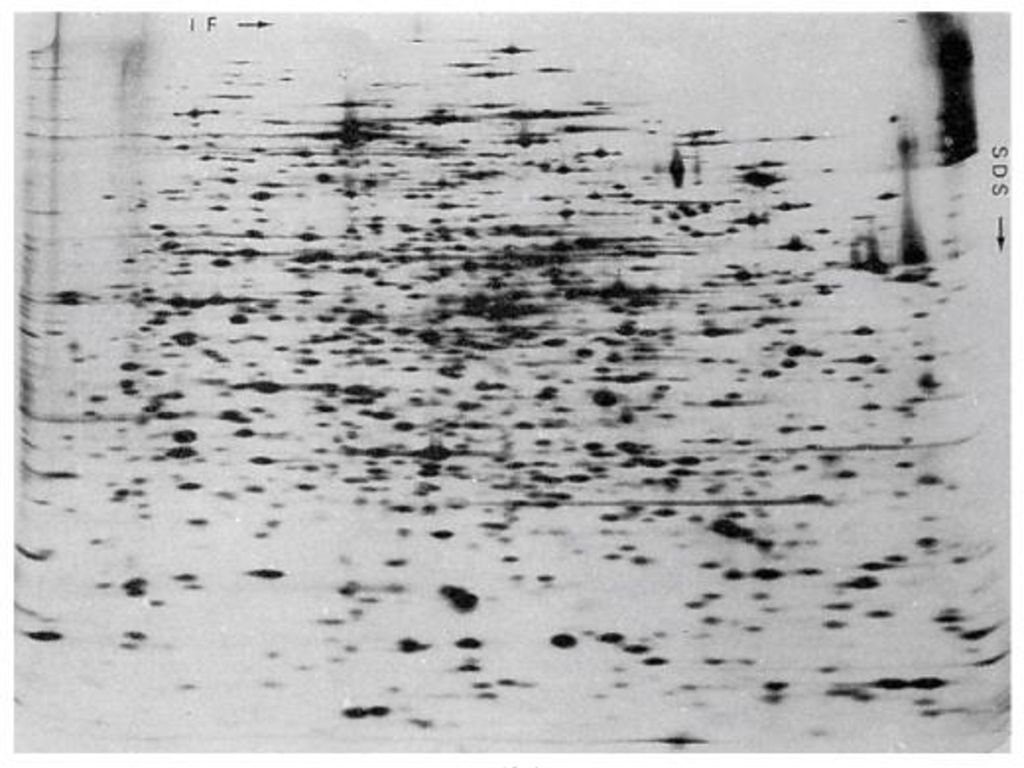
双向电泳技术 1975年首次建立了等电聚焦及SDS-聚丙烯酰胺凝胶双向电泳技术,该方法依赖于蛋白质分子的等电点和相对分子质量的差异。 目前,双向电泳的分辨率达到每块胶10000个蛋白白点。

81
蛋白质印迹法 蛋白质印迹法(Western blotting),又称为免疫印迹法(immunoblotting),用于鉴定蛋白质,其原理是根据被测蛋白质能与特异性抗体结合的特性和该蛋白质的相对分子质量。
,又称为免疫印迹法(immunoblotting),用于鉴定蛋白质,其原理是根据被测蛋白质能与特异性抗体结合的特性和该蛋白质的相对分子质量。")
82
图5-28 Western Blotting示意图

84
5.6.3 蛋白质的质谱分析 生物质谱法用于鉴定电泳分离后的蛋白质。错误率:低于百万分之一。 工作原理:
蛋白质的质谱分析 生物质谱法用于鉴定电泳分离后的蛋白质。错误率:低于百万分之一。 工作原理: 蛋白质酶解成肽段与基质混合 激光轰击、喷射分析 达到检测器的时间由质量/电荷比决定质谱图比较分析:通过比较肽质量指纹图谱与数据库中不同肽段的理论分子质量来寻找。

85
图5-30 MALDI-TOF分子质谱仪原理

Similar presentations



:核蛋白体组成成分 转移 RNA ( tRNA ):转运氨基酸 信使 RNA ( mRNA ):蛋白质合成模板 不均一核 RNA ( hnRNA ):成熟 mRNA 的前体 小核 RNA ( snRNA ):>")


DNA、RNA及蛋白质操作技术.>")













>")

 A.细菌的遗传物质主要是DNA B.病毒的遗传物质主要是RNA>")