Download presentation
Presentation is loading. Please wait.

1
荧光定量PCR 刘 兵

2
目录 一、背景知识 二、荧光定量PCR概述 三、荧光定量PCR的主要原理 四、荧光定量PCR定量的理论依据 五、荧光定量PCR定量的理论模式

3
一、背景知识 1、1985 年,美国Perkin-Elmer Cetus 公司人类遗传研究室Mullis 等发明了具有划时代意义的聚合酶链反应(Polymerase chain reaction, PCR),使人们梦寐以求的体外无限扩增核酸片段的愿望成为现实。 (陈旭等,2010)
")
4
2、1992 年,Higuchi最早提出了实时PCR 的设想,它的基本思想是利用荧光染料EB(溴化乙锭)可以插入到双链核酸中受激发光的性质,在PCR反应的退火或延伸时检测掺入到双链核酸中EB的含量就能实时监控PCR反应的进程,根据PCR反应的数学函数关系,结合相应的算法,通过加入标准品的方法,就可以对待测样品中的目标基因进行准确定量。(陈旭等,2010)
可以插入到双链核酸中受激发光的性质,在PCR反应的退火或延伸时检测掺入到双链核酸中EB的含量就能实时监控PCR反应的进程,根据PCR反应的数学函数关系,结合相应的算法,通过加入标准品的方法,就可以对待测样品中的目标基因进行准确定量。(陈旭等,2010)")
5
3、到1995 年,美国PE公司研制成功具有革命性意义的荧光定量PCR技术,融合了PCR高灵敏性、DNA杂交的高特异性和光谱技术的高精确定量等优点,直接探测PCR过程中荧光信号的变化,以获得特定区段扩增产物定量的结果,不需要PCR后处理或电泳检测,完全闭管操作(整个过程中仅有加入样品的一次开盖),一举克服了常规PCR技术的诸多难题。 (陈旭等,2010)
,一举克服了常规PCR技术的诸多难题。 (陈旭等,2010)")
6
二、荧光定量PCR概述 (1)在荧光定量PCR 反应中, 引入了一种荧光化学物质,随着PCR 反应的进行,PCR 反应产物不断累计,荧光信号强度也等比例增加。每经过一个循环,收集一个荧光强度信号,这样就可以通过荧光强度变化监测产物量的变化。从而得到一条荧光扩增曲线。(朱捷等,2009)
在荧光定量PCR 反应中, 引入了一种荧光化学物质,随着PCR 反应的进行,PCR 反应产物不断累计,荧光信号强度也等比例增加。每经过一个循环,收集一个荧光强度信号,这样就可以通过荧光强度变化监测产物量的变化。从而得到一条荧光扩增曲线。(朱捷等,2009)")
7
(2)、荧光扩增曲线包括3 个阶段:基线期,扩增期和平台期。只有在荧光信号扩增期,PCR 产物量的对数值与起始模板量之间存在线性关系,所以可以在这个阶段进行定量分析。(朱捷等,2009)
(3)、为了便于对所检测样本进行比较,在荧光定量PCR 反应的指数期,需要设定一定荧光信号的阈值,一般这个阈值是以PCR反应的前15 个循环的荧光信号作为荧光本地信号, 荧光阈值的缺省设置是3~15 个循环的荧光信号的标准偏差的10 倍。 (朱捷等,2009)
、为了便于对所检测样本进行比较,在荧光定量PCR 反应的指数期,需要设定一定荧光信号的阈值,一般这个阈值是以PCR反应的前15 个循环的荧光信号作为荧光本地信号, 荧光阈值的缺省设置是3~15 个循环的荧光信号的标准偏差的10 倍。 (朱捷等,2009)")
8
(4)、如果检测到荧光信号超过阈值被认为是真正的信号,它可以用于定义样本的阈值循环数(Cycle Threshold,Ct)。每个模板的Ct 值与该模板的起始拷贝数存在线性关系。在PCR 过程中可以连续不断地检测反应体系中荧光信号的变化。当信号增强到荧光阈值时,Ct 值被记录下来。(朱捷等,2009) 注:Ct值是指样品管的荧光信号达到某一固定阈值的PCR反应循环数。在PCR循环过程中, 即荧光信号开始由本底进入指数增长阶段的拐点所对应的循环次数。在实际操作中, 这个时刻也就是每个反应管内的荧光信号到达设定的域值的时刻, 可见Ct值取决于阈值。(赵焕英等,2007)
")
9
(5)、起始拷贝数越多,Ct 值越小。利用己知起始拷贝数的标准品可作出标准曲线,只要获得未知样品的Ct 值, 即可从标准曲线上计算出该样品的起始拷贝数。 (朱捷等,2009)
、起始拷贝数越多,Ct 值越小。利用己知起始拷贝数的标准品可作出标准曲线,只要获得未知样品的Ct 值, 即可从标准曲线上计算出该样品的起始拷贝数。 (朱捷等,2009)")
10
三、荧光定量PCR的主要原理 荧光共振能量转移,外来的光源激发供体荧光染料发出荧光,当其发射波长与受体荧光染料的吸收波长部分重叠,且两者的距离很近(约10~100埃)时,受体荧光染料吸收供体荧光传递的能量而激发出另一波长的荧光,同时将供体荧光淬灭。
时,受体荧光染料吸收供体荧光传递的能量而激发出另一波长的荧光,同时将供体荧光淬灭。")
11
四、荧光定量PCR定量的理论依据 (1)、特定的待扩增基因片段起始含量越大,则指数扩增过程越短,当扩增速率趋于稳定后,则无论原来样品中起始模板含量多少,最终扩增片段的含量通常是一样的。 (2)、理想的扩增结果:Y=X×2n 其中Y代表扩增产物量,X代表PCR反应体系中的原始模板数,n为扩增次数。
、特定的待扩增基因片段起始含量越大,则指数扩增过程越短,当扩增速率趋于稳定后,则无论原来样品中起始模板含量多少,最终扩增片段的含量通常是一样的。 (2)、理想的扩增结果:Y=X×2n 其中Y代表扩增产物量,X代表PCR反应体系中的原始模板数,n为扩增次数。")
12
(3)、理论上PCR扩增效率为100%,但实际上:DNA的每一次复制都不完全,实际应为: Y=X×(1+E)n,其中E代表扩增效率,通常E≤1通常X在1~105拷贝、循环次数n ≤30时,E相对稳定,原始模板以相对固定的指数形式增加,适合定量分析,这也就是所谓的指数期;随着循环次数的增加,E值逐渐减少,Y呈非固定的指数形式增加,最后进入平台期。
、理论上PCR扩增效率为100%,但实际上:DNA的每一次复制都不完全,实际应为: Y=X×(1+E)n,其中E代表扩增效率,通常E≤1通常X在1~105拷贝、循环次数n ≤30时,E相对稳定,原始模板以相对固定的指数形式增加,适合定量分析,这也就是所谓的指数期;随着循环次数的增加,E值逐渐减少,Y呈非固定的指数形式增加,最后进入平台期。")
13
五、荧光定量PCR定量的理论模 式 (1)、PCR是对原始待测模板核酸的一个扩增过程,任何干扰PCR指数扩增的因素都会影响扩增产物的量,使得PCR扩增终产物的数量与原始模板数量之间没有一个固定的比例关系,通过检测扩增终产物很难对原始模板进行准确的定量。
、PCR是对原始待测模板核酸的一个扩增过程,任何干扰PCR指数扩增的因素都会影响扩增产物的量,使得PCR扩增终产物的数量与原始模板数量之间没有一个固定的比例关系,通过检测扩增终产物很难对原始模板进行准确的定量。")
14
(2)、荧光定量PCR能相对准确的定量PCR,
PCR扩增通式:① Tn=T0(1+E)n ② Tn=Tn-1(1+E) 注:[0<E<1] 其中E表示扩增速率,n为循环数,Tn表示在n个周期后PCR产物的量,Tn-1表示在n-1个周期后PCR产物的量,T0为原始模板数量。
n. ② Tn=Tn-1(1+E) 注:[0<E<1] 其中E表示扩增速率,n为循环数,Tn表示在n个周期后PCR产物的量,Tn-1表示在n-1个周期后PCR产物的量,T0为原始模板数量。")
15
CP=-1/lg(1+E)*lgT0+lgK/lg(1+E)
(3)、设定PCR到达指数扩增期时,产生一定的荧光(荧光依赖于某一循环扩增产物的总量,即特定的拷贝数K,大约在1010左右)高于背景为仪器所识别,此时的循环数为CP(crossing point),也就是K=T0 (1+E)CP,取对数得: lgK=lgT0+CP*lg(1+E) CP=-1/lg(1+E)*lgT0+lgK/lg(1+E) 由于对一特定的PCR而言,E与K均为常数,故上式为循环数CP对原始模板拷贝数的对数lgT0一次方程,其标准形式y=kx+b,该直线的斜率为 -1/lg(1+E),在横坐标上的截距为 lgK/lg(1+E),依据此作PCR标准曲线。
、设定PCR到达指数扩增期时,产生一定的荧光(荧光依赖于某一循环扩增产物的总量,即特定的拷贝数K,大约在1010左右)高于背景为仪器所识别,此时的循环数为CP(crossing point),也就是K=T0 (1+E)CP,取对数得: lgK=lgT0+CP*lg(1+E) CP=-1/lg(1+E)*lgT0+lgK/lg(1+E) 由于对一特定的PCR而言,E与K均为常数,故上式为循环数CP对原始模板拷贝数的对数lgT0一次方程,其标准形式y=kx+b,该直线的斜率为. -1/lg(1+E),在横坐标上的截距为 lgK/lg(1+E),依据此作PCR标准曲线。")
16
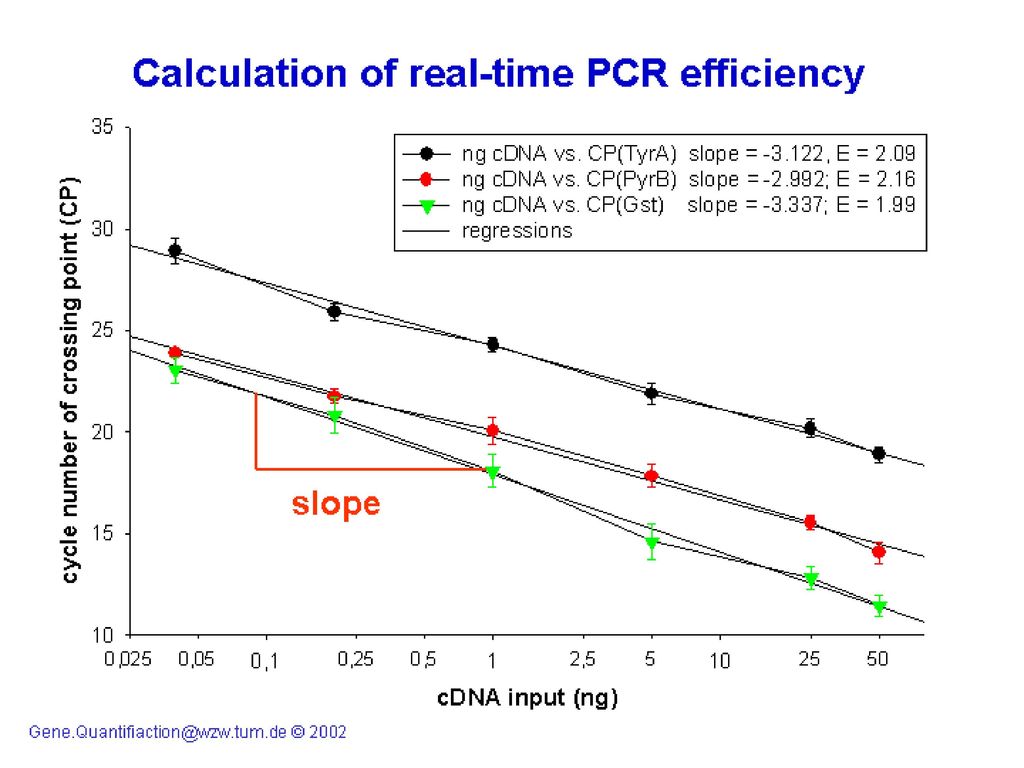
单管实时荧光定量RT-PCR的标准曲线

17
目前荧光定量PCR均采用外标定量 (1)、由于标准曲线的斜率(Slope)为- 1/lg(1+E),可知E=10-1/Slope-1 ,如从标准曲线中知道Slope=-3.337,则E=101/ =0.99,也有人将(1+E)直接视为扩增效率E,则E=10-1/Slope,求得的E值均在1—2之间,有时也有大于2的结果。
、由于标准曲线的斜率(Slope)为- 1/lg(1+E),可知E=10-1/Slope-1 ,如从标准曲线中知道Slope=-3.337,则E=101/ =0.99,也有人将(1+E)直接视为扩增效率E,则E=10-1/Slope,求得的E值均在1—2之间,有时也有大于2的结果。")
19
(2)、也可直接根据系列稀释外标在该次实时荧光PCR的结果来大略分析扩增效率的高低,例如系列10倍稀释外标在PCR扩增时有N2=N0En2, N3=(10*N0)En3,在扩增到指数期时荧光值相同则代表此点的终末拷贝数相同,即N2=N3,En2-n3=10,取对数得到 ⊿nlgE=1,E=101/⊿n,E=2,⊿n=3.32;E=1.8,⊿n=3.91。反之亦然。

21
荧光定量PCR使用外标定量的原因: A、内标对实时荧光定量PCR的影响 若在待测样品中加入已知起始拷贝数的内标,则PCR反应变为双重PCR,双重PCR反应中存在两种模板之间的干扰和竞争,尤其当两种模板的起始拷贝数相差比较大时,这种竞争会表现得更为显著。但由于待测样品的起始拷贝数是未知的,所以无法加入合适数量的已知模板作为内标。也正是这个原因,传统定量方法虽然加入内标,但仍然只是一种半定量的方法。

22
B、荧光定量PCR无需内标定量 实时荧光定量PCR无需内标是建立在两个基础之上的: (1)Ct值的高度重现性 PCR循环在到达Ct值所在的循环数时,刚刚进入真正的指数扩增期(对数期),此时微小误差尚未放大,因此Ct值的重现性极好,即同一模板不同时间扩增或同一时间不同管内扩增,得到的Ct值是恒定的。
,此时微小误差尚未放大,因此Ct值的重现性极好,即同一模板不同时间扩增或同一时间不同管内扩增,得到的Ct值是恒定的。")
23
(2)Ct值与起始模板的线性关系 由于Ct值与起始模板的对数存在线性关系,可利用标准曲线对未知样品进行定量测定,因此,实时荧光定量PCR是一种采用外标准曲线定量的方法。
Ct值与起始模板的线性关系 由于Ct值与起始模板的对数存在线性关系,可利用标准曲线对未知样品进行定量测定,因此,实时荧光定量PCR是一种采用外标准曲线定量的方法。")
24
六、 荧光定量PCR的分类 实时荧光定量PCR 包括探针类和染料类两种, 探针类是利用与靶序列特异杂交的探针来指示扩增产物的增加。染料类如SYBR Green I则是利用与双链DNA 小沟结合发光的理化特征指示扩增产物的增加。

25
1、双链DNA(dsDNA)结合染料 (1)、Sybr Green I 检测模式 SYBR Green I 是一种能与双链DNA 结合发光的荧光染料。其与双链DNA结合后,荧光大大增强。因此, SYBR Green I的荧光信号强度与双链DNA的数量有关,可以根据荧光信号检测出PCR体系存在的双链DNA数量。SYBR Green I 的最大吸收波长约为497nm,发射波长最大约为520nm。PCR扩增程序一般为94℃~55℃~72℃三步法,40个循环。

26
SYBR Green I 的缺点:由于SYBR Green I没有特异性,不能识别特定的双链,只要是双链就会结合发光,对PCR反应中的非特异性扩增或引物二聚体也会产生荧光,通常本底较高,所以在临床上使用可能会有假阳性发生。 SYBR Green I的优点:SYBR Green I的优点是因为其缺点产生,由于它能所有的双链DNA相结合,所以对不同模板不需特别定制不同的特异性探针,通用性较好,并且价格相对较低。这对科研是很有利的,因此国内外在科研中使用比较普遍。

27
SYBR GREEN I 工作原理

28
(2)、LC Green TM I 与SYBRGreen I相比, 该染料是一种饱和结合染料, 能够完全结合dsDNA分子,可直接加入PCR反应体系中,高浓度的LC Green TM I不会抑制PCR反应。已有文献报道在PCR反应体系中加入LC Green TM I, 使用一对引物, 一个不作标记的探针, 结合常规融解曲线或仅使用一对引物, PCR 结束后通过分析融解曲线的形状和融解温度(Tm值)的大小, 对纯合子和杂合子以及寡核苷酸序列多态性(SNP)进行鉴别。LC Green TM I染料法简便、快速、精确性高, 预期将会有很好的应用前景。(赵焕英等,2007)
的大小, 对纯合子和杂合子以及寡核苷酸序列多态性(SNP)进行鉴别。LC Green TM I染料法简便、快速、精确性高, 预期将会有很好的应用前景。(赵焕英等,2007)")
29
2、荧光标记探针 (1)、 水解探针模式(Taqman) TaqMan 探针是一种寡核苷酸探针,荧光基团连接在探针的5,末端,而淬灭剂则在3,末端。当探针与靶序列配对时,荧光基团发射的荧光因与3,端的淬灭剂接近而被淬灭。在进行延伸反应时,聚合酶的5,外切酶活性将探针切断,使得荧光基因与淬灭剂分离,发射荧光。一分子的产物生成就伴随着一分子的荧光信号的产生。随着扩增循环次数的增加,释放出来的荧光基团不断积累。因此Taqman探针检测的是积累荧光。PCR扩增程序通常是:94℃ ~ 60 ℃ 40 个循环。常用的荧光基团是 FAM,TET,VIC,HEX。

30
Taqman探针工作原理

31
(2)、分子信标 分子信标是一种呈发夹结构的茎环双标记寡核苷酸探针,两端的核酸序列互补配对, 因此标记在一端的荧光基团与标记在另一端的淬灭基团紧紧靠近。荧光基团被激发后产生的光子被淬灭剂淬灭,由荧光基团产生的能量以红外而不是可见光形式释放出来。
、分子信标 分子信标是一种呈发夹结构的茎环双标记寡核苷酸探针,两端的核酸序列互补配对, 因此标记在一端的荧光基团与标记在另一端的淬灭基团紧紧靠近。荧光基团被激发后产生的光子被淬灭剂淬灭,由荧光基团产生的能量以红外而不是可见光形式释放出来。")
32
分子信标的茎环结构中,环一般为15-30个核苷酸长,并与目标序列互补;茎一般5-7个核苷酸长,并相互配对形成茎的结构。在复性温度下,因为模板不存在时形成茎环结构,当加热变性会互补配对的茎环双链解开,如果有模板存在环序列将与模板配对。与模板配对后,分子信标将成链状而非发夹状,使得荧光基团与淬灭剂分开。当荧光基团被激发时,因淬灭作用被解除,发出激发光子。由于是酶切作用的存在,分子信标也是积累荧光。常用的荧光基团:FAM ,Texas Red 。PCR扩增程序通常是:94~55~72 ℃三步法,40个循环。

33
分子信标工作原理

34
(3)、 FRET探针 FRET探针又称双杂交探针, FRET探针由两条相邻探针组成,在一条探针的5,端标记FAM荧光基团,另一探针的3,端标记Red640荧光基团。当复性时,探针结合在模板上,FAM基团和Red640基团相邻,激发FAM产生的荧光,作为Red640基团的激发光被吸收,使 Red640 发出波长为640的荧光。当变性时,探针游离,两基团距离远,不能产生640波长的荧光。由于FRET探针是靠近发光,所以检测信号是实时信号,非累积信号。常用的荧光基团是:LC-Red640,LC-Red705。

35
FRET工作原理

36
几种探针的比较:

37
七、传统的定量PCR的难点 (1)如何确定PCR正处于线性扩增范围内(只有在此范围内PCR产物信号才与初始模板的拷贝数成比例)。
(2)一旦线性扩增范围确定以后,如何找到一个合适的方法检测结果。为了保证线性,研究者要扩增一系列梯度稀释的cDNA或者对每个基因的扩增循环数作适当的调整;有的研究者加一个竞争性模板,有的做限制性的稀释梯度分析,或者加一个PCR模拟(mimic)反应,即用相似的引物与目标产物共扩增,而扩增产物的检测通常在跑胶以后通过染料、放射活性或者探针进行检测。
一旦线性扩增范围确定以后,如何找到一个合适的方法检测结果。为了保证线性,研究者要扩增一系列梯度稀释的cDNA或者对每个基因的扩增循环数作适当的调整;有的研究者加一个竞争性模板,有的做限制性的稀释梯度分析,或者加一个PCR模拟(mimic)反应,即用相似的引物与目标产物共扩增,而扩增产物的检测通常在跑胶以后通过染料、放射活性或者探针进行检测。")
38
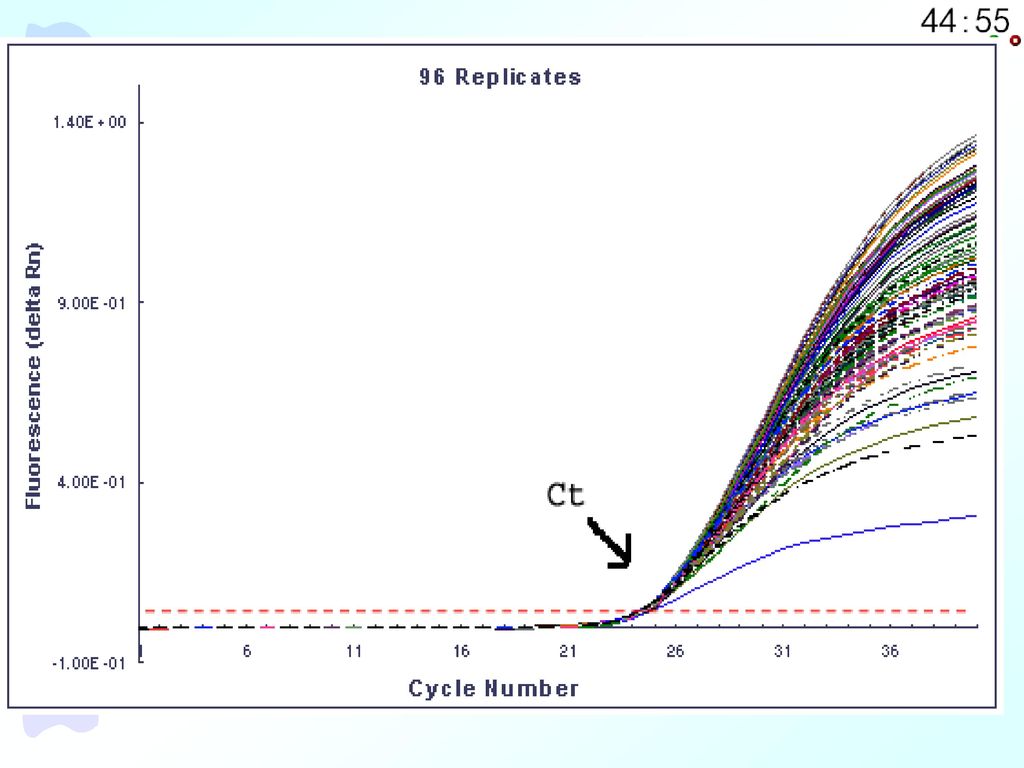
(3)大多数是终点检测,而PCR经过对数期扩增到达平台期时,检测重现性极差。同一个模板在96孔PCR仪上做96次重复实验,所得结果有很大差异(如下图所示),因此无法直接从终点产物量推算出起始模板量;虽然加入内标后,可部分消除终产物定量所造成的不准确性。但即使如此,传统的定量方法也都只能算作半定量、粗略定量的方法,所以传统定量PCR的缺点是:劳动强度大、定量不准确、重复性差。
大多数是终点检测,而PCR经过对数期扩增到达平台期时,检测重现性极差。同一个模板在96孔PCR仪上做96次重复实验,所得结果有很大差异(如下图所示),因此无法直接从终点产物量推算出起始模板量;虽然加入内标后,可部分消除终产物定量所造成的不准确性。但即使如此,传统的定量方法也都只能算作半定量、粗略定量的方法,所以传统定量PCR的缺点是:劳动强度大、定量不准确、重复性差。")
40
八、 荧光定量PCR的优点 (1)、特异性好,使用特异性探针对定量分子进行识别,具有很高的准确性。同时,靶序列由引物和探针双重控制,特异性好、假阳性低。 (2)、灵敏度高,荧光PCR检测技术是综合了PCR技术、荧光标记技术、激光技术、数码显象技术为一体的技术,因此它的检测灵敏度很高。 (3)、线性关系好、线性范围宽,由于荧光信号的产生和每次扩增产物成一一对应的关系,通过荧光信号的检测可以直接对产物进行定量;定量范围可在0-1010拷贝/毫升。 (4)、操作简单、安全、自动化程度高、防污染。扩增和检测可以在同一管内检测,不需要开盖,不易污染;同时扩增和检测一步完成,不需要后期处理,不再需要担心放射性污染。 (5)、速度快、高通量,可在2-3小时完成96个样品的定量分析。
、线性关系好、线性范围宽,由于荧光信号的产生和每次扩增产物成一一对应的关系,通过荧光信号的检测可以直接对产物进行定量;定量范围可在0-1010拷贝/毫升。 (4)、操作简单、安全、自动化程度高、防污染。扩增和检测可以在同一管内检测,不需要开盖,不易污染;同时扩增和检测一步完成,不需要后期处理,不再需要担心放射性污染。 (5)、速度快、高通量,可在2-3小时完成96个样品的定量分析。")
41
九、怎样设计荧光定量PCR实验方案 1、基因序列的查找 2、引物、探针的设计 3、荧光定量PCR方法的确定 4、引物探针合成标记
5、标准品的制备 6、反应液的配制 7、反应条件的设定 8、基数的设定、数据分析

42
引物设计通常遵守如下原则: A、引物与模板的序列要紧密互补。 B、引物与引物之间避免形成稳定的二聚体或发夹 结构。 C、引物不能在模板的非目的位点引发DNA 聚合反应(即错配)。 D、引物长度通常在20-25bp,Tm值在55-65℃,GC含量在40%-60%,产物大小在 bp之间。 E、引物的3’端避免使用碱基A;引物3’端避免出现3 个以上连续相同的碱基。 F、为避免基因组的扩增,引物设计最好能跨两个外显子。

43
探针设计通常遵守如下原则: A、探针位置尽可能地靠近上游引物。 B、探针长度通常在20-30bp,Tm值在65-70℃,通常比引物高5-10℃,GC含量在40%-70%。 C、探针的5’端应避免使用碱基G。 D、整条探针中,碱基C的含量要明显高于 G的含量。

44
十、荧光定量PCR 技术在科研中的应用(朱捷等,2009)
(1)DNA 或RNA 的定量检测应用 包括病原微生物或病毒含量的检测,转基因动植物转基因拷贝数的检测,RNAi 基因失活率的检测等。实时荧光定量PCR 技术可以快速、准确定量测出
DNA 或RNA 的定量检测应用. 包括病原微生物或病毒含量的检测,转基因动植物转基因拷贝数的检测,RNAi 基因失活率的检测等。实时荧光定量PCR 技术可以快速、准确定量测出.")
45
(2)、基因表达研究的检测应用 荧光定量PCR 技术对mRNA 水平的检测比Northern 杂交、RT-PCR 等定量方法要方便、快速、准确得多。荧光定量PCR技术可以对不同组织、不同处理之间(如药物处理、物理处理和化学处理等)、不同发育阶段基因表达差异进行检测,检测各基因在组织细胞中的表达丰度, 分析基因的表达调控、mRNA 的表达模式等研究。
、不同发育阶段基因表达差异进行检测,检测各基因在组织细胞中的表达丰度, 分析基因的表达调控、mRNA 的表达模式等研究。")
46
(3)、食品安全中的检测应用 人们在日常生活中食品是否安全,进出口食品和粮食是否含有危险或潜在危险的成分,都可以用荧光定量PCR 技术进行快速检测,荧光定量PCR 方法是国内外进行转基因成分含量检测的常用方法。例如, 利用RQ -PCR 技术定量检测谷物基因来控制缺乏麦麸的婴儿食物。
、食品安全中的检测应用 人们在日常生活中食品是否安全,进出口食品和粮食是否含有危险或潜在危险的成分,都可以用荧光定量PCR 技术进行快速检测,荧光定量PCR 方法是国内外进行转基因成分含量检测的常用方法。例如, 利用RQ -PCR 技术定量检测谷物基因来控制缺乏麦麸的婴儿食物。")
47
(4)、医学及药物开发中的检测应用 荧光定量PCR 技术广泛的应用于临床及其药物方面,目前已经在肿瘤研究,乙肝诊断,病毒检测,动物疾病检测与防御,药物疗效的评价与考核等方面,为临床理论研究、治疗疾病和药物开发提供了客观的物质基础。
、医学及药物开发中的检测应用 荧光定量PCR 技术广泛的应用于临床及其药物方面,目前已经在肿瘤研究,乙肝诊断,病毒检测,动物疾病检测与防御,药物疗效的评价与考核等方面,为临床理论研究、治疗疾病和药物开发提供了客观的物质基础。")
48
(5)、遗传学及SNPs 分析上的应用 荧光定量PCR 技术还可以用在点突变分析、等位基因分析、DNA 甲基化检测、单核苷酸多态性(SNP)分析及特异突变基因检测等。根据TaqMan 探针法在扩增之前处理DNA, 然后用特异的引物和探针可以区分甲基化和非甲基化的DNA。SNP 分析从根本上来说是确定一对染色体的每种基因的两个拷贝,结合荧光定量PCR 可以快速的检测SNP 结果。
分析及特异突变基因检测等。根据TaqMan 探针法在扩增之前处理DNA, 然后用特异的引物和探针可以区分甲基化和非甲基化的DNA。SNP 分析从根本上来说是确定一对染色体的每种基因的两个拷贝,结合荧光定量PCR 可以快速的检测SNP 结果。")
49
谢谢!

Similar presentations

的作用而发生一系列质的 改变,形成一种异常的增生。这种异常 增生一旦形成,即使外界至病因素停止 作用,也还能继续自然发展,这说明肿 瘤是整体性疾病的一种局部表现。>")
















不同生物间DNA分子的结构基本相同; (2)不同生物进行基因表达时都遵循“中心法则”; (3)所有生物共用一套遗传密码.>")

