Download presentation
Presentation is loading. Please wait.

1
第二章 高分子的聚集态结构

2
第二章 高分子的聚集态结构 1、高分子的聚集态结构是指高分子链之间的排列和堆砌结构,也称为超分子结构。
第二章 高分子的聚集态结构 1、高分子的聚集态结构是指高分子链之间的排列和堆砌结构,也称为超分子结构。 高分子的链结构是决定高聚物基本性质的主要因素,而高分子的聚集态结构是决定高聚物本体性质的主要因素。对于实际应用中的高聚物材料或制品,其使用性能直接决定于在加工成型过程中形成的聚集态结构。 链结构只是间接影响高聚物材料的性能,而聚集态结构才是直接影响其性能的因素。 链结构是在高分子的合成过程中形成的,而聚集态结构是在高分子加工、成型过程中形成的。

3
高分子凝聚态是指高分子链之间的几何排列和堆砌状态
晶态 固体 聚集态为物质的物理状态 相态为物质的热力学状态 液态 液体 气态 气体 晶态 固体 非晶态 液晶态 高分子凝聚态是指高分子链之间的几何排列和堆砌状态 取向结构 织态结构 决定 决定 高分子的凝聚态结构 聚合物的基本性能特点 材料的性能 获得 得到 预定材料结构 预定材料性能 控制成型加工条件

4
第一节 高聚物分子间的作用力 2.1.1 范德华力与氢键 分子间的作用力包括范德华力(静电力、诱导力和色散力)和氢键。
第一节 高聚物分子间的作用力 范德华力与氢键 分子间的作用力包括范德华力(静电力、诱导力和色散力)和氢键。 内聚能密度 1、高分子的聚集态只有固态和液态,而没有气态,说明高分子的分子间力超过了组成它的化学键的键能。因为物质只有在破坏掉其分子间力时才会变为气态,可此时化学键已经被破坏而分解。 2、高聚物分子间力的大小采用内聚能或内聚能密度表示。 3、内聚能:把一摩尔液体或固体分子移到其分子间力范围之外缩需要的能量。 单位体积的内聚能叫内聚能密度 ⊿E= ⊿Hv-RT CED= ⊿E/V 对于低分子化合物,其内聚能近似等于恒容蒸发热或升华热。而高聚物不能汽化,不能直接测定内聚能(密度),只能估计。
和氢键。 内聚能密度. 1、高分子的聚集态只有固态和液态,而没有气态,说明高分子的分子间力超过了组成它的化学键的键能。因为物质只有在破坏掉其分子间力时才会变为气态,可此时化学键已经被破坏而分解。 2、高聚物分子间力的大小采用内聚能或内聚能密度表示。 3、内聚能:把一摩尔液体或固体分子移到其分子间力范围之外缩需要的能量。 单位体积的内聚能叫内聚能密度. ⊿E= ⊿Hv-RT CED= ⊿E/V. 对于低分子化合物,其内聚能近似等于恒容蒸发热或升华热。而高聚物不能汽化,不能直接测定内聚能(密度),只能估计。")
5
表 线型高聚物的内聚能密度 高聚物 内聚能密度(兆焦/米3) PE 259 PMMA 347 PIB 272 PVAc 368 天然橡胶
表 线型高聚物的内聚能密度 高聚物 内聚能密度(兆焦/米3) PE 259 PMMA 347 PIB 272 PVAc 368 天然橡胶 280 PVC 381 PB 276 PET 477 丁苯橡胶 Nylon-66 774 PS 305 PAN 992
 PE PMMA PIB PVAc 天然橡胶 PVC PB PET 丁苯橡胶. Nylon PS PAN")
6
内聚能密度与高聚物的物理性能存在明显的对应关系,内聚能密度在290兆焦/米3以下的高聚物均为非极性聚合物,分子间力小,分子链柔顺性好,富有弹性,可用作橡胶(PE例外,它易于结晶而失去弹性,只能作为塑料);高于420的高聚物,分子链上有强极性基团,或分子间能形成氢键,机械强度高,耐热,分子链结构规整,易于结晶取向,强度高,可以作纤维,其间的高聚物适合作塑料。
;高于420的高聚物,分子链上有强极性基团,或分子间能形成氢键,机械强度高,耐热,分子链结构规整,易于结晶取向,强度高,可以作纤维,其间的高聚物适合作塑料。")
7
2.2 聚合物的晶态结构Crystalline structure
熔体结晶 方 法 高分子链本身具有必要的规整结构 高分子结晶,形成晶体 玻璃体结晶 溶液结晶 适宜的温度,外力等条件 Debye ring X射线衍射花样 X-ray patterns or Debye crystallogram 结晶聚合物的重要实验证据 X射线衍射曲线 X-ray diffraction

8
X-ray instrument Soller slit – Soller狭缝 2 Scan

9
无规聚丙烯和等规聚丙烯的X-ray图 无规聚丙烯 弥散圆 等规聚丙烯 弥散圆和衍射环共存

10
2.2.1 高聚物结晶的形态学 1、单晶 高聚物的单晶通常只能在极稀的溶液中(0.01~0.1%)缓慢结晶时生成的,在电镜下可以直接观察到它们是具有规则几何形状的薄片状晶体。
缓慢结晶时生成的,在电镜下可以直接观察到它们是具有规则几何形状的薄片状晶体。")
11
PE单晶 螺旋生长 稀溶液,慢降温

12
影响单晶生长的因素 1)溶液浓度:溶液浓度必须足够稀,使溶液中的高分子能够彼此分离,避免因分子链相互缠接。通常0.01-0.1%浓度。
2)结晶温度:结晶温度必须足够高,或者过冷程度要小(结晶熔点与结晶温度之差),使结晶速度足够慢,保证分子链的充分排列。一般过冷温度20-30K。 3)其它:溶剂的性质对结晶影响也很大。通常采用热力学上的不良溶剂(溶解能力较差的溶剂)。分子量对结晶也有影响,在同一温度下,高分子倾向于按分子量由大到小的顺序结晶。 要注意,高分子单晶是由溶液中生长的片状晶体的总称,并非结晶学意义上的真正单晶 ..1
结晶温度:结晶温度必须足够高,或者过冷程度要小(结晶熔点与结晶温度之差),使结晶速度足够慢,保证分子链的充分排列。一般过冷温度20-30K。 3)其它:溶剂的性质对结晶影响也很大。通常采用热力学上的不良溶剂(溶解能力较差的溶剂)。分子量对结晶也有影响,在同一温度下,高分子倾向于按分子量由大到小的顺序结晶。 要注意,高分子单晶是由溶液中生长的片状晶体的总称,并非结晶学意义上的真正单晶")
13
2)球晶是由一个晶核开始,以相同的生长速率同时向空间各个方向放射生长形成的。
2、球晶 1) 球晶是高聚物结晶的最常见形式,当结晶性的高聚物从浓溶液中析出,或从熔体冷却结晶时,都倾向于生成这种结晶,它呈圆球型。在偏光显微镜下观察,球晶呈现特征的黑十字消光图案。 2)球晶是由一个晶核开始,以相同的生长速率同时向空间各个方向放射生长形成的。
 球晶是高聚物结晶的最常见形式,当结晶性的高聚物从浓溶液中析出,或从熔体冷却结晶时,都倾向于生成这种结晶,它呈圆球型。在偏光显微镜下观察,球晶呈现特征的黑十字消光图案。 2)球晶是由一个晶核开始,以相同的生长速率同时向空间各个方向放射生长形成的。")
14
3)在晶核较少,而且球晶较小时,呈球形;当晶核较多,并继续生长扩大后,他们之间会出现非球形的界面。同时成核并以相同速度生长的球晶界面是一个平面,而不同时间生长或生长速度不同的球晶界面是回转双曲面。
4)当球晶生长一直进行到充满整个空间时,球晶将成为不规则的多面体。 5)在球晶的偏光显微镜研究中,在特征的黑十字消光图案上,还重叠着明暗相间的消光同心圆环(P45),这是径向发射的晶片缎带状协同扭转的结果。
当球晶生长一直进行到充满整个空间时,球晶将成为不规则的多面体。 5)在球晶的偏光显微镜研究中,在特征的黑十字消光图案上,还重叠着明暗相间的消光同心圆环(P45),这是径向发射的晶片缎带状协同扭转的结果。")
15
Maltese Cross in Isotactic Polystyrene
偏光显微镜观察

16
Polarized-light microscopy

17
偏光显微镜下球晶的生长 0s 30s 60s 90s 120s

18
The growth of spherulites

19
两种球晶

20
1)树枝状晶:溶液中析出,浓度较高、结晶温度较低或分子量过大时生成。
3、其它结晶形式 1)树枝状晶:溶液中析出,浓度较高、结晶温度较低或分子量过大时生成。 2)孪晶:在孪生片晶的不同部分具有结晶学上不同取向的晶胞的一类晶体。一般从溶液中生长,在低分子量高聚物中较常见。 3)伸直链片晶:由完全伸展的分子链平行规整排列而成的片状晶体,其晶片厚度可与分子链的伸展长度相当。形成于熔体加压结晶过程。
树枝状晶:溶液中析出,浓度较高、结晶温度较低或分子量过大时生成。 2)孪晶:在孪生片晶的不同部分具有结晶学上不同取向的晶胞的一类晶体。一般从溶液中生长,在低分子量高聚物中较常见。 3)伸直链片晶:由完全伸展的分子链平行规整排列而成的片状晶体,其晶片厚度可与分子链的伸展长度相当。形成于熔体加压结晶过程。")
21
shish-kebab structure
串晶 Folded chain Extended chain

22
4)纤维状晶和串晶:当存在流动场时,高分子链的构象发生畸变。成为伸展的形式,并沿流动方向平行排列,适当条件下成核结晶而成纤维状晶,其长度可以不受分子链长度的限制。分子链的取向平行于纤维轴。
在适当条件下,在纤维状晶的表面上外延生长许多片状附晶,形成一种类似于串珠似结构的特殊结晶形态,即串晶。 这种流动或应变诱发结晶,与实际生产过程中高聚物的结晶过程更为接近。

23
2.2.2 高分子在结晶中的构象和晶胞 1、具体内容不作要求,但要注意以下几点:
1)结晶中高分子的构象决定于分子内和分子间两种力的作用。除少量分子间力(如氢键)较大的高聚物(如尼龙)外,分子间力的影响是有限的,常常可以忽略不计。 2)高聚物结晶时不会出现立方晶格,因为立方晶格是各向同性的。而高分子链在结晶时都只能采取使其主链的中心轴互相平行的方式排列。与主链中心轴平行的方向就是晶胞的主轴,在该方向上有化学键,而在空间的其它方向只有分子间力。在分子间力作用下,分子链只能靠近到链外原子或取代基接近到范德华距离为度,这就产生了各向异性。 3)由于高分子的长链结构,链上的原子有共价键,结晶时链段并不能充分自由运动,必定妨碍规整排列,从而造成晶格缺陷,也就是高分子结晶一般不完整,常出现准晶、非晶区。
结晶中高分子的构象决定于分子内和分子间两种力的作用。除少量分子间力(如氢键)较大的高聚物(如尼龙)外,分子间力的影响是有限的,常常可以忽略不计。 2)高聚物结晶时不会出现立方晶格,因为立方晶格是各向同性的。而高分子链在结晶时都只能采取使其主链的中心轴互相平行的方式排列。与主链中心轴平行的方向就是晶胞的主轴,在该方向上有化学键,而在空间的其它方向只有分子间力。在分子间力作用下,分子链只能靠近到链外原子或取代基接近到范德华距离为度,这就产生了各向异性。 3)由于高分子的长链结构,链上的原子有共价键,结晶时链段并不能充分自由运动,必定妨碍规整排列,从而造成晶格缺陷,也就是高分子结晶一般不完整,常出现准晶、非晶区。")
24
第三节 高分子的聚集态结构模型 理论假设 不作要求

25
第四节 高聚物的结晶过程 1、链的对称性 高分子链的结构对称性越高越容易结晶。PE和PTFE对称性好,最容易结晶,但将PE 氯化后(CPE)对称性受到破坏,便失去了原有的结晶能力。 2、链的规整性 无规聚合物一般不易结晶,如自由基聚合得到PS、PMMA、PVAc不能结晶,而用定向聚合得到的聚合物则具有结晶能力。(聚α-烯烃) 在二烯类聚合物中,由于存在顺反异构,如果几何构型无规排列,则不能结晶,通过定向聚合得到的全顺式或全反式结构的聚合物,则获得结晶能力,顺式聚合物的结晶能力小于反式聚合物。
 在二烯类聚合物中,由于存在顺反异构,如果几何构型无规排列,则不能结晶,通过定向聚合得到的全顺式或全反式结构的聚合物,则获得结晶能力,顺式聚合物的结晶能力小于反式聚合物。")
26
几个值得注意的例外 1、自由基聚合得到的聚三氟氯乙烯,主链上有不对称碳原子,而又不是等规聚合物,却具有相当强的结晶能力,最高结晶度可达90%。这是由于氯原子与氟原子体积相差不大,不妨碍分子链做规整的堆积。类似于PTFE。 2、无规PVAc不能结晶,但由它水解得到的PVA能结晶,原因在于羟基的体积不大,而又具有较强的极性的缘故。 3、无规PVC具有微弱的结晶能力。原因在于氯原子电负性较大,分子链上的氯原子相互排斥彼此错开排列,形成类似于间同立构的结构,有利于结晶。

27
共聚物的结晶能力 1、无规共聚物通常会破坏链的对称性和规整性,从而使结晶能力降低甚至完全丧失。但是如果两种高聚单元的均聚物有相同类型的结晶结构,那么共聚物也能结晶。如果两种共聚单元的均聚物有不同的结晶结构,那么在一种组分占优势时,共聚物是可以结晶的,含量少的结构单元作为缺陷存在于另一种均聚物的结晶结构中。但是在某些中间组成时,结晶能力大大减弱。甚至不能结晶,如乙丙共聚物。 2、嵌段共聚物的各嵌段基本保持相对独立性,能结晶的嵌段形成自己的晶区。

28
其它结构因素 1、链的柔顺性:一定的链的柔顺性是结晶时链段向结晶表面扩散和排列所必需的。例如链柔顺性好PE结晶能力高,而主链上含苯环的PBET柔性下降,结晶能力较低,而主链上苯环密度更高的聚碳酸酯,链的柔性更差,结晶能力更差。 2、支化使链的对称性和规整性降低,降低结晶能力。例如高压法制备的PE的结晶能力小于低压线形PE。 为何高压PE叫做LDPE,而低压PE叫做HDPE。 3、交联大大限制了链的活动性,随着交联度的增加,结晶能力下降。 4、分子间力也往往使链的柔顺性降低,影响结晶能力。但分子间能形成氢键时,则有利于结晶结构的稳定。

29
2.4.2 结晶速度及其测定方法 1、 高聚物的结晶过程与小分子类似,也包括晶核的形成和晶粒的生长两个步骤,因此结晶速度也包括成核速度、结晶生长速度和由它们共同决定的结晶总速度。 2、测定方法包括: 成核速度: 用偏光显微镜、电镜直接观察单位时间内的成核数目。 结晶生长速度: 用偏光显微镜、小角激光光散射法测定球晶半径随时间的增大速度。 结晶总速度: 用膨胀计法、光学解偏振法测定结晶过程进行到一半时的时间,以其倒数作为结晶总速度。

30
膨胀计法设备简单,但热平衡时间较长,起始时间不易测准,难以研究结晶速度较快的过程。 …………………
膨胀计法研究高聚物结晶过程 该法利用高聚物结晶过程中发生体积收缩来研究结晶过程。具体方法: 将高聚物与惰性液体装入膨胀计中,加热到共聚物的熔点以上,使高聚物全部成为非晶熔体,然后将膨胀计移入恒温槽中,高聚物开始恒温结晶,观察膨胀计毛细管内液体的的高度随时间的变化,便可以考察结晶进行的情况。以h0、h、ht分别代表膨胀计的起始、最终和t时间时的读数,将( ht - h )/( h0 - h )对t作图,得到如图反S型曲线。 由曲线可以看出,结晶过程开始体积收缩慢,过一段时间后加快,之后又逐渐慢下来,最后体积收缩变得非常缓慢,这时结晶速度的衡量发生困难,变化终点所需的时间也不明确,然而体积收缩一半所需的时间可以准确测量,而且此时体积变化的速度较大,时间测量误差小,因此常用其倒数表示结晶速度,t1/2 膨胀计法设备简单,但热平衡时间较长,起始时间不易测准,难以研究结晶速度较快的过程。 ………… ………………… t1/2
/( h0 - h )对t作图,得到如图反S型曲线。 由曲线可以看出,结晶过程开始体积收缩慢,过一段时间后加快,之后又逐渐慢下来,最后体积收缩变得非常缓慢,这时结晶速度的衡量发生困难,变化终点所需的时间也不明确,然而体积收缩一半所需的时间可以准确测量,而且此时体积变化的速度较大,时间测量误差小,因此常用其倒数表示结晶速度,t1/2. 膨胀计法设备简单,但热平衡时间较长,起始时间不易测准,难以研究结晶速度较快的过程。 ………… ………………… t1/2.")
31
此外,尚有光学解偏振法、偏光显微镜法和小角激光光散射法,不作要求 膨胀计法研究高聚物结晶情况中应用的液体应具备什么条件?
此外,尚有光学解偏振法、偏光显微镜法和小角激光光散射法,不作要求 膨胀计法研究高聚物结晶情况中应用的液体应具备什么条件?

32
2.4.4结晶速度与温度的关系 高聚物本体结晶温度范围都在其玻璃化温度与熔点之间。在某一适当温度下,结晶速度出现极大值。 Tmax = 0.63Tm Tg-18.5 Tmax = 0.85Tm

33
虽然,目前还不能从理论上全面比较不同高聚物的结晶速度,但可以证明:
2.4.5影响结晶速度的其它因素 分子结构的差别是决定高聚物结晶速度的根本原因。 虽然,目前还不能从理论上全面比较不同高聚物的结晶速度,但可以证明: 链的结构越简单、对称性越高、链的立体规整性越好、取代基的空间位阻越小、链的柔顺性越大,则结晶速度越大 同一种高聚物,分子量大,结晶速度慢,因此为了达到同样的结晶度,分子量大的高聚物热处理时间越长。

34
有些杂质可以促进结晶的形成,在结晶过程中起到晶核的作用,被称为成核剂。加入成核剂可以加快结晶速度,并使球晶变小。这在工业生产中已广泛被采用。
杂质的存在对结晶过程影响很大。 有些杂质能够阻碍结晶的进行,如惰性稀释剂可降低结晶分子浓度,从而降低结晶速度。例如在等规聚合物中加入相同化学组成的无规聚合物,可以使结晶速度降低到所需要的水平。这一现象常被用于研究那些结晶速度过快的聚合物的结晶行为。 有些杂质可以促进结晶的形成,在结晶过程中起到晶核的作用,被称为成核剂。加入成核剂可以加快结晶速度,并使球晶变小。这在工业生产中已广泛被采用。

35
补充内容: PP成核剂和透明剂 传统的成核剂大多是芳族羧酸酯,如苯甲酸钠等。 透明剂是高档的成核剂 山梨醇缩醛 有机磷酸酯 松香酸盐等

36
常见PP制品 加入透明剂的PP制品

37
第五节 结晶对高聚物物理机械性能的影响 结晶度概念及其测定方法 结晶高聚物中总是包含晶区和非晶区两部分,结晶度如下: fcw=Wc/(Wc+Wa)×100% fcv=Vc/(Vc+Va)×100% W表示重量,V表示体积,下标c表示结晶,a表示非晶 用文字表述结晶度概念

38
高聚物的晶区与非晶区的界限不明确,这给准确缺定晶区和非晶区含量带来麻烦用不同方法测定的结晶度,一般要表明测定方法,因为各种方法对晶区和非晶区的定义是不同的。(举例:P77)
简介密度梯度法测结晶度的方法

39
2.5.2 结晶度大小对高聚物性能的影响 同一种单体,以不同的聚合方法或不同的成型条件可以制得结晶或不结晶的高分子材料。
举例:PP:制备方法不同 PVA:加工方法不同,结晶度不同,普通的只有1/3结晶,遇热水溶解,而230度热处理85分钟,可提高到65%,在90度的热水中溶解很少,定向聚合得到等规聚乙烯醇,结晶度很高,不经缩醛化反应,便可以作纤维。 PE:不溶于烃类溶剂,因为结晶度高。结晶可以提高耐热性和耐溶剂侵蚀性。 对于塑料和纤维,通常希望它们有合适的结晶度,对于橡胶则不希望其有结晶性,结晶会使橡胶硬化而失去弹性。例如汽车轮胎在北方的冬天有时会因为结晶而破裂。

40
结晶对高聚物性能的影响 1、力学性能 当高聚物的非晶区位于橡胶态(高弹态)时,模量随结晶度的提高而增加,硬度增高。 结晶度提高,抗冲击强度降低。在玻璃化温度以上,结晶度增加,分子间作用力增加,抗张强度提高,但断裂伸长减小;在玻璃化温度以下,高聚物随结晶度增加而变脆,抗张强度下降。 在玻璃化温度以上,微晶起物理交联点的作用,使链的滑移减小,蠕变和应力松弛降低。

41
2、密度和光学性质: 晶区密度大于非晶区,因此随结晶度增加密度增加,大量实验表明,结晶和非晶密度的比值约为1.13。 ρc/ ρa= 1.13,因此测得某一样品的密度,即可粗略估计其结晶度ρ = ρa ×(1+0.13fcv) 物质的折光率与密度有关,因此高聚物中晶区与非晶区折光率不同,光线通过时在晶区界面上发生折射和反射,不能直接通过。因此两相并存的结晶高聚物通常呈乳白色,不透明,如PE、PA、PTFE等,结晶度减小,透明度增加,完全非晶的聚合物是透明的,如PMMA、PS等。

42
PE POM

43
PTFE制品

44
PMMA

45
PC

46
但是,有的高聚物晶区密度和非晶区密度差别很小,或者晶体尺寸比可见光波长还小,此时结晶并不影响高聚物的透明性。
例如,聚4-甲基-1-戊烯,分子链上有较大的侧基,使其结晶排列不紧密,两相密度很接近,是透明高聚物。对于许多结晶高聚物,可以设法减小结晶尺寸,例如等规PP加工时加入成核剂,透明度改善。

47
3、热性能: 对于塑料,非晶或结晶度小时,其最高使用温度是玻璃化温度。当结晶度达到40%后,晶区互相连接,成为连续相,在玻璃化温度以上,仍不软化,其最高使用温度可提高到熔点。 4、其他性能 由于结晶使分子链紧密堆积,它能更好阻挡各种试剂的渗入,及其对气体、液体、蒸汽等的渗透性、化学反应活性等性能。

48
2.5.3 结晶高聚物的加工条件-结构-性质的关系 不同结晶度的高聚物具有不同的物理机械性能,因此就具有不同的用途。 1、聚三氟氯乙烯,mp210ºC。如果缓慢冷却,结晶度可达85-90%,用淬火的方法可使其结晶度降到35-40%。(性能见P81表2-18) 聚三氟氯乙烯耐腐蚀性好,常被涂于化工容器表面防腐,结晶度的控制很重要,结晶度高的产品,硬而脆,耐冲击性能不强,为提高其韧性,淬火处理,获得低结晶度涂层。 另外,它还用于制造防腐零件。但不能长期在120ºC以上使用,因为,它在120ºC以下结晶速度很小,超过该温度,结晶速度增加。

49
2、PE 作薄膜用时要求透明性和韧性好,结晶度应低,作为塑料,要求有足够的强度和刚性,结晶度要高,因此在选用材料时,要…?(原料、加工)
")
50
3、聚酯纤维的结晶度对性能影响 当融化的聚酯从喷丝头出来后,若能迅速而均匀地冷却,其结晶速度慢,结晶度低,结晶度低的聚酯纤维在牵伸时能达到的牵引倍数就比较大,使高分子链的取向性好,整个纤维的性能比较均匀,所以要严格控制纺丝吹风窗的温度。

51
2.5.4 分子量等因素对结晶高聚物的聚集态结构的影响
分子量等因素对结晶高聚物的聚集态结构的影响 分子量大于某一数值以上的样品,结晶度随分子量的增加而单调下降,一直到很高的分子量,最后趋于某一极限值。 其他,不作要求。

52
在通常的升温速度下,结晶高聚物熔融过程的体积(或比热)对温度的曲线见P85。
第六节 结晶热力学 2.6.1 结晶高聚物的熔融与熔点 在通常的升温速度下,结晶高聚物熔融过程的体积(或比热)对温度的曲线见P85。 结晶高聚物的熔融过程与小分子结晶既相似又有差别,相似之处在于也发生热力学函数(体积、比热等)的突变,但不象小分子那样发生在约0.2ºC左右的窄温度范围内,而是有一个较宽的温度范围,这范围叫熔限。在熔限内,发生边熔融边升温的现象,而不象小分子那样,几乎保持在一个温度下,直到晶相全部熔融为止。
对温度的曲线见P85。 结晶高聚物的熔融过程与小分子结晶既相似又有差别,相似之处在于也发生热力学函数(体积、比热等)的突变,但不象小分子那样发生在约0.2ºC左右的窄温度范围内,而是有一个较宽的温度范围,这范围叫熔限。在熔限内,发生边熔融边升温的现象,而不象小分子那样,几乎保持在一个温度下,直到晶相全部熔融为止。")
53
改变升温方式,如温升1度,便保温直至体积不变。表明结晶高聚物的熔融过程与小分子一样,主要发生在3-4ºC范围内,与小分子晶体的熔化过程只有量的差别,没有本质区别。熔融终点处对应的温度为高聚物的熔点。
结晶高聚物出现的边熔融边升温现象的解释: 结晶高聚物中含有完善程度不同的结晶的缘故,不完善的晶体在较低的温度下熔融,便出现较宽的熔限。而在缓慢升温情况下,可以使不完善的晶体充分熔融再结晶为完善的晶体(充分再结晶的机会),因为结晶和熔融是可逆的。
,因为结晶和熔融是可逆的。")
54
原因:在较低的温度下结晶时,分子链的活动能力较差,形成的晶体较不完善,完善的程度差别也较差,自然这样的晶体将在较低的温度下熔融,熔限也较宽。
2.6.2结晶温度对熔点的影响 结晶温度越低,熔点越低,熔限越宽 结晶温度越高,熔点越高,熔限越窄。 原因:在较低的温度下结晶时,分子链的活动能力较差,形成的晶体较不完善,完善的程度差别也较差,自然这样的晶体将在较低的温度下熔融,熔限也较宽。

55
2.6.3 晶片厚度与熔点的关系 晶片厚度越大,熔点越高。 2.6.4 拉伸对高聚物熔点的影响
对于结晶高聚物,牵伸能帮助高聚物结晶,提高结晶度,同时也提高了熔点

56
2.6.5 高分子链结构对熔点的影响 1、等规烯类聚合物 当PE的的次甲基规则地带上烷基取代基时,即等规聚α-烯烃,由于主链内旋转位阻增加,分子链的柔顺性降低,熔点升高。但当正烷基侧链长度增加时,影响了链间的紧密堆积,使熔点下降(P89、90),但四碳以后,重新出现有序性的堆砌,熔点回升。
,但四碳以后,重新出现有序性的堆砌,熔点回升。 .")
57
2、脂肪族聚酯、尼龙和聚氨酯 这几类聚合物的熔点随重复单元长度的变化呈现统一的总趋势,重复单元长度增加,逐渐接近PE的熔点,几类聚合物比较,其熔点升高的顺序为: POLYESTER 、PE、聚氨酯、PA、PU

59
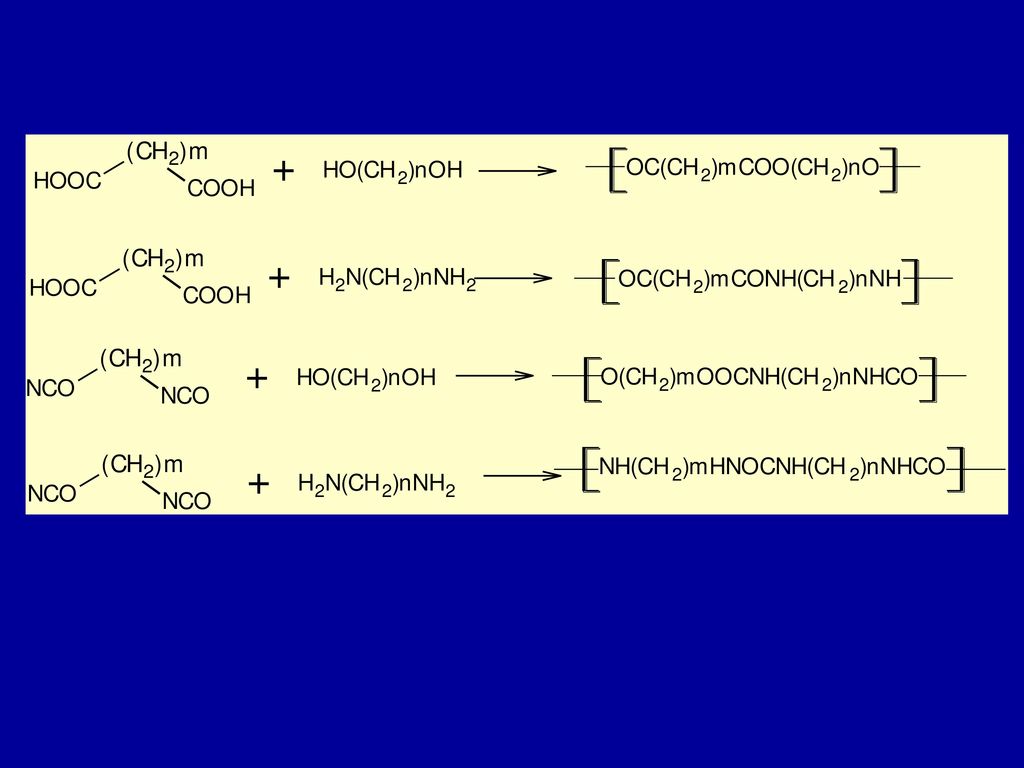
补充材料:聚氨酯 聚氨酯生产原料:二异氰酸酯、多元醇、多元胺 应用面最宽的高分子材料

62
在主链上含环状结构或共轭结构的聚合物都使链的刚性大大增加,因此都具有较高的熔点。如苯环、萘环等。环状结构密度越大,熔点越高。
3、主链含苯环或其他刚性结构的高聚物 在主链上含环状结构或共轭结构的聚合物都使链的刚性大大增加,因此都具有较高的熔点。如苯环、萘环等。环状结构密度越大,熔点越高。 对位芳族高聚物的熔点比相应的间位的熔点高。涤纶的熔点为280ºC,而其间位聚合物熔点为240ºC。

63
4、其他聚合物 PTFE具有很高的熔点327ºC,在其结晶熔融后,接近其分解温度时还没有观察到流动现象,因此,它不能用加工热塑性塑料的方法进行加工。原因在于它的构象几乎接近棒状。 二烯类1,4-聚合物都具有较低的熔点,顺式结构聚合物的熔点比反式更低。

64
不沾锅涂料的主要成分为PTFE, 其本身无毒,但是其在高温分解时 会放出有毒的小分子 关键是温度的控制
补充内容 不沾锅涂料的毒性问题 不沾锅涂料的主要成分为PTFE, 其本身无毒,但是其在高温分解时 会放出有毒的小分子 关键是温度的控制

65
2.6.6共聚物的熔点 当结晶聚合物的单体与另一单体进行共聚时,如果,该单体本身不能结晶,或虽能结晶,但不能进入原结晶聚合物的晶格,形成共晶,则生成共聚物的结晶行为将发生变化,结晶熔点Tm与原结晶聚合物的平衡熔点Tm0的关系如下(P代表共聚物中结晶单元相增长的几率,R是气体常数, ΔHu是每摩尔重复单元的熔融热。) 1/Tm-1/Tm0=(-R/ΔHu)lnP 以上关系表明,共聚物的熔点与组成没有直接的关系,而是决定于共聚物的序列分布情况。
 1/Tm-1/Tm0=(-R/ΔHu)lnP. 以上关系表明,共聚物的熔点与组成没有直接的关系,而是决定于共聚物的序列分布情况。")
66
1、无规共聚物 P≡XA,(结晶单元的摩尔分数),随非晶共聚物单元的增加,熔点降低,直到一个适当的组成,这时共聚物两个组分的熔点相同,达到低共熔点。
,随非晶共聚物单元的增加,熔点降低,直到一个适当的组成,这时共聚物两个组分的熔点相同,达到低共熔点。")
67
典型无规共聚物熔点与组成关系图 I 对苯二甲酸、己二酸与乙二醇共聚物 II对苯二甲酸、癸二酸与乙二醇共聚物
IV己二酸、己二胺与己内酰胺共聚物

68
对于嵌段共聚物,P >XA,有时甚至接近于1,该类共聚物大多只有轻微的相对于其均聚物的熔点降低。
2、嵌段共聚物 对于嵌段共聚物,P >XA,有时甚至接近于1,该类共聚物大多只有轻微的相对于其均聚物的熔点降低。

69
3、交替共聚物, P <XA ,熔点将具有明显的降低。 因此,具有相同组成的共聚物,因序列分布不同,其熔点具有很大的差别。
在实际应用中,嵌段和无规共聚均可用于有目的地降低熔点。

70
对苯二甲酸/己二酸乙二醇共聚物的熔点

71
2.6.7杂质对高聚物熔点的影响 1、杂质使熔点降低。 1/Tm-1/Tm0=(-R/ΔHu)lnaA,aA为结晶组分的活度,当杂质浓度很低时,它等于结晶部分的摩尔分数XA。
lnaA,aA为结晶组分的活度,当杂质浓度很低时,它等于结晶部分的摩尔分数XA。")
72
2、低分子稀释剂(增塑剂、未聚合单体及其他可溶性添加剂):
1/Tm-1/Tm0=(R/ΔHu)(Vu/V1)(φ1-χ1 φ12) 式中Vu、V1、φ1、χ1分别为高分子重复单元和小分子稀释剂的摩尔体积,体积分数和高分子与稀释剂的作用参数。对于溶解能力很好的稀释剂, χ1可为负数,随着溶解能力的降低,其值增大,良溶剂使高聚物熔点降低更多
(Vu/V1)(φ1-χ1 φ12) 式中Vu、V1、φ1、χ1分别为高分子重复单元和小分子稀释剂的摩尔体积,体积分数和高分子与稀释剂的作用参数。对于溶解能力很好的稀释剂, χ1可为负数,随着溶解能力的降低,其值增大,良溶剂使高聚物熔点降低更多.")
73
3、端基可以当作杂质处理,它也会引起结晶熔点的降低。
1/Tm-1/Tm0=2/Pn(R/ΔHu),Pn为数均聚合度。
,Pn为数均聚合度。")
74
第七节 高聚物的取向态结构 2.7.1 高聚物的取向现象 当线形高分子充分伸展的时候,其长度为其宽度的102~104倍,这种结构上悬殊的不对称性,使其在某些情况下很容易沿某特定方向作占优势的平行排列,这就是取向。

75
取向(orientation):在外力作用下,分子链沿外力方向平行排列。聚合物的取向现象包括分子链、链段的取向以及结晶聚合物的晶片等沿特定方向的择优排列。
:在外力作用下,分子链沿外力方向平行排列。聚合物的取向现象包括分子链、链段的取向以及结晶聚合物的晶片等沿特定方向的择优排列。")
76
取向与结晶的比较 取向态与结晶态都与高分子的有序性有关,但其有序程度是不同的,取向态是一维或二维在一定程度上的有序,而结晶态则是三维有序的。

77
取向造成各向异性 未取向的聚合物材料是各向同性的,即各个方向上的性能相同(isotropic)。而取向后的聚合物材料是各向异性的(anisotropic),即方向不同,性能也不同。 双轴拉伸或吹塑的薄膜 纤维 聚合物取向方法 熔融挤出的管材和棒材

78
无定形聚合物 Amorphous polymer
链段取向 非晶区 Amorphous region 取向单元 链段取向 晶态聚合物 Crystalline polymer 晶区 Crystal region 球晶变形,晶片 倾斜、滑移、取向

79
取向对高聚物性能的影响 1、力学性能 抗张强度和挠曲疲劳强度在取向方向上显著增加,而与取向方向垂直的方向上则降低,冲击强度和断裂伸长也发生相应变化。 2、光学性能 取向造成高分子出现双折射现象,即在平行和垂直于取向方向上的折射率发生了变化,光学各向异性以其差值表示。 Δn=n//- n┴ 。 3、使用温度 取向使玻璃化温度提高,结晶高聚物的密度和结晶度也提高,因此提高了高分子的使用温度。

80
取向高分子材料的分类 1、单轴取向 常见为纤维,纤维成型时,从喷丝孔出来时已经部分取向,再经过牵伸若干倍,使进一步取向,强度提高。

81
2、双轴取向 常见为薄膜材料。单轴取向薄膜只在薄膜平面的某一方向上具有高强度,而垂直取向方向上强度却降低,实际应用中,薄膜将在该方向上首先被破坏。而双轴取向的薄膜,分子链平行于薄膜的任意方向,这薄膜在平面上各向同性。

82
Different Types of Orientation
Uniaxial orientation Biaxial orientation

83
Biaxial Orientation 双轴取向:一般在两个垂直方向施加外力。如薄膜双轴拉伸,使分子链取向平行薄膜平面的任意方向。在薄膜平面的各方向的性能相近,但薄膜平面与平面之间易剥离。 薄膜的双轴拉伸取向 薄膜挤压吹塑机

84
2.7.2 高聚物的取向机理 1、高分子的取向分链段取向和整个分子链取向两种。 2、链段取向可以通过单键的内旋转实现,这种取向过程在高弹态下就能进行。 3、分子链的取向需要高分子各链段协同运动才能实现,只有处于粘流态才能实现。

85
4、不同取向的高分子的性能是不同的,分子链取向的材料具有明显的各向异性,而链段取向的材料就不明显。
5、取向过程是链段运动的过程,需要克服分子间的作用力,因此完成取向需要一定时间,两种取向方式需要克服阻力的大小不同,因此,其取向速度也不相同。在外力作用下,首先发生链段取向,然后才是整个分子的取向。

86
6、取向与热运动是相反的过程,前者是分子的有序化过程,必须依靠外力作用实现;后者是自发过程,使分子趋向无序。取向状态是热力学上非平衡态。在高弹态,拉伸可以使链段取向,但一旦外力除去,就自发解取向;在粘流态下,分子链可以取向,外力消失后,分子也要解取向。为了维持取向状态,必须使温度迅速降到玻璃化温度以下,将分子和链段的运动“冻结”起来,这种状态仍是热力学非平衡态,只有相对稳定性,时间长了,尤其是温度升高或高聚物被溶剂溶胀后,就会解取向,取向过程快的,解取向也快。

87
7、结晶高聚物的取向,除了非晶区的链段和分子链会发生取向以外,还可能发生晶粒的取向。在外力作用下,晶粒将沿外力方向择优取向。具体取向机理尚存在争论。

88
2.7.3 取向度及其测定方法 1、表征聚合物取向程度的物理量 2、一般以取向函数F表示:
F=1/2(3cos2θ-1),式中θ是分子链主轴与取向方向间的夹角。对于理想的单轴取向,在链取向方向上θ=0°, cos2θ=1,F=1;在垂直于链取向方向上, θ=90°, cos2θ=0,F=-0.5;完全无规取向的, F=0, cos2θ=1/3, θ=54°44' 。 3、测定取向度的方法很多,声波传播法、光学双折射法、广角X射线衍射法、红外二色相法以及偏振荧光法等 4、声波传播法:声波在高分子中的传播速度在沿着分子主链方向上传播速度要比垂直于链的方向上快得多,如果以Cu和C分别代表无规取向的高聚物及待测试样中的声速,则F=1-( Cu /C)2。
,式中θ是分子链主轴与取向方向间的夹角。对于理想的单轴取向,在链取向方向上θ=0°, cos2θ=1,F=1;在垂直于链取向方向上, θ=90°, cos2θ=0,F=-0.5;完全无规取向的, F=0, cos2θ=1/3, θ=54°44 。 3、测定取向度的方法很多,声波传播法、光学双折射法、广角X射线衍射法、红外二色相法以及偏振荧光法等. 4、声波传播法:声波在高分子中的传播速度在沿着分子主链方向上传播速度要比垂直于链的方向上快得多,如果以Cu和C分别代表无规取向的高聚物及待测试样中的声速,则F=1-( Cu /C)2。")
89
2.7.4 取向研究的应用 1、合成纤维材料的牵伸 完全分子链取向的纤维强度高,但断裂伸长小,脆性大,一般要求有10~20%的断裂伸长,需要进行热处理,使链段解取向。 工业上采用慢的取向过程,使整个分子链取向,达到高强度,再用快的过程使链段解取向,以获得高弹性。例:黏胶纤维的加工过程。 热处理(热定型)目的:除了以上目的,还可减少纤维的沸水收缩率。 各种纤维要求的取向程度是不同的。分子刚性程度、能否结晶,分子间相互作用力等决定。
目的:除了以上目的,还可减少纤维的沸水收缩率。 各种纤维要求的取向程度是不同的。分子刚性程度、能否结晶,分子间相互作用力等决定。")
90
为了制备稳定的具有相当弹性的取向纤维或薄膜,要解决为使取向态稳定就必须减少分子的活动能力,而要维持弹性必须增加分子的活动能力之间的矛盾。对于非晶高分子,无论分子刚性还是柔性,都无法解决这一矛盾。只有用结晶聚合物才能解决这一矛盾。

91
2、薄膜材料的双轴取向 对于板材进行双向拉伸(相互垂直方向),对于管材进行吹塑工艺,同时在纵向进行拉伸。 3、塑料制品的取向 塑料制件往往形状复杂,无法进行拉伸取向,但取向对塑料制品也具有重要意义。塑料要求具有良好的取向能力。 对于外型比较简单的薄壁塑料制品,利用取向来提高强度的例子很多,例如战斗机的透明机舱罩、安全帽的生产、中空制品的吹塑成型等。

92
第八节 高聚物的液晶态结构 液晶态结构 1、定义:某些物质的结晶在熔融或溶解之后,虽然失去固态物质的刚性,获得液态物质的流动性,却仍然部分保持晶态物质的有序排列,从而在物理性质上呈现各向异性,形成一种兼有晶体和液体部分性质的过度状态,称为液晶态,这种物质称为液晶。 2、液晶分子通常具有刚性的分子结构,分子的长度和宽度之比R 1,呈棒状或似棒状构象,同时还应具有在液态维持分子的某种有序排列所必需的凝聚力(晶原或介原),一般与对位苯撑、强极性基团、高度可极化基团或氢键相连;此外液晶的流动性还要求分子结构上必须有一定的柔性部分。 3、液晶按形成形式分为热致型液晶和溶致型液晶两类。 4、根据液晶晶原部分结构分为筷、碟、碗型 5、根据分子排列的形式和有序性的不同,分为近晶型、向列型和胆甾型三种。
,一般与对位苯撑、强极性基团、高度可极化基团或氢键相连;此外液晶的流动性还要求分子结构上必须有一定的柔性部分。 3、液晶按形成形式分为热致型液晶和溶致型液晶两类。 4、根据液晶晶原部分结构分为筷、碟、碗型. 5、根据分子排列的形式和有序性的不同,分为近晶型、向列型和胆甾型三种。")
93
液晶—有序流动的液体 液晶的特点——同时具有流动性和光学各向异性
晶体三维有序 液晶 液态的无序

94
(2)The history of liquid crystal 液晶的历史
1888年 F. Reinitzer (F. Reinitzer; Monatsh. Chem., 9, 421, 1888 ), a botanist from Austria, and after that, O. Lehmann, a German crystal researcher, verified the optical anisotropy of the crystal. Lehmann proposed to call it the Fliessende krystalle , in English that is Liquid Crystal or simplified as LC. 60年代,美国杜邦公司(Du Pont’s)先后推出了PBA(聚苯甲酰胺)及Kevelar纤维(PPTA, 聚对苯二甲酰对苯二胺),标志了液晶研究的工业化发展的开始。 70~80年代,出现了诸如Xydar(美国Darton公司 1984年), Vectra(美国Calanese公司 1985年)等一系列商用型热致型液晶。
, a botanist from Austria, and after that, O. Lehmann, a German crystal researcher, verified the optical anisotropy of the crystal. Lehmann proposed to call it the Fliessende krystalle , in English that is Liquid Crystal or simplified as LC. 60年代,美国杜邦公司(Du Pont’s)先后推出了PBA(聚苯甲酰胺)及Kevelar纤维(PPTA, 聚对苯二甲酰对苯二胺),标志了液晶研究的工业化发展的开始。 70~80年代,出现了诸如Xydar(美国Darton公司 1984年), Vectra(美国Calanese公司 1985年)等一系列商用型热致型液晶。")
95
Pierre-Gilles de Gennes (1932-
The Nobel Prize in Physics 1991 "for discovering that methods developed for studying order phenomena in simple systems can be generalized to more complex forms of matter, in particular to liquid crystals and polymers" France

96
(棒状)向列相液晶(Nemactic)
向列相液晶(Nemactic)")
97
(棒状)近晶相A液晶(Smectic A)
近晶相A液晶(Smectic A)")
98
(棒状)近晶相C(Smectic C)
近晶相C(Smectic C)")
99
胆甾相液晶(Ch)
")
100
盘状液晶 Discotic Discotic Nematic –DN Discotic hexegonal disordered – Dhd
Discotic hexegonal ordered – Dho Discotic hexegonal disordered – Dhd

101
按液晶基元所在位置分类 主链型液晶 侧链型液晶

102
侧链液晶 主链液晶 串型 介晶基元位于分子主链的高分子称为主链型液晶高分子。 介晶基元位于分子侧基的高分子称为侧链型液晶高分子。 腰接侧链型
组合式

103
按液晶形成条件分类 溶致液晶:在某一温度下,因加入溶剂而呈现液晶态的物质 ---- 核酸,蛋白质,芳族聚酰胺PBA, PPTA (Kevlar) 和聚芳杂环PBZT, PBO 热致液晶:通过加热而形成液晶态的物质 ---- 共聚酯, 聚芳酯Xydar, Vector, Rodrum 感应液晶:外场(力,电,磁,光等)作用下进入液晶态的物质 ---- PE under high pressure 流致液晶:通过施加流动场而形成液晶态的物质 ----聚对苯二甲酰对氨基苯甲酰肼
作用下进入液晶态的物质 ---- PE under high pressure. 流致液晶:通过施加流动场而形成液晶态的物质 ----聚对苯二甲酰对氨基苯甲酰肼.")
104
(5) Characterization and application of liquid crystal polymer液晶的表征和应用
Polarized-light microscopy 偏光显微镜 DSC – Differential scanning clarometry示差扫描量热法 液晶态的表征 XRD - X-ray diffraction X射线衍射 液晶原位增强聚合 液晶的应用 液晶显示 LCD- Liquid crystal display 液晶纺丝:在低牵伸倍数下获得高度取向、高性能纤维。

105
1、高分子液晶按其晶原所处的位置分为主链液晶(主链由液晶原和柔性链节组成)和侧链液晶(主链柔性,侧链刚性晶原)两类。
2.8.2 高分子液晶的结构和性能 1、高分子液晶按其晶原所处的位置分为主链液晶(主链由液晶原和柔性链节组成)和侧链液晶(主链柔性,侧链刚性晶原)两类。 2、筷型高分子液晶最常见,碗型高分子液晶尚未合成 3、高分子液晶的研究始于五十年代,早期侧重于多肽的溶液,此后发现芳香族尼龙的溶液向列型液晶。 4、高分子液晶的性质不同于一般高分子,尤其是其独特的流动特性。
和侧链液晶(主链柔性,侧链刚性晶原)两类。 2、筷型高分子液晶最常见,碗型高分子液晶尚未合成. 3、高分子液晶的研究始于五十年代,早期侧重于多肽的溶液,此后发现芳香族尼龙的溶液向列型液晶。 4、高分子液晶的性质不同于一般高分子,尤其是其独特的流动特性。")
106
高分子液晶的流动特性

108
高分子液晶在剪切力作用下的流动特性

109
2.8.7 高分子液晶的应用 1、液晶显示技术 将高分子液晶薄膜夹在两块导电玻璃板之间,在施加适当电压的点上,高分子变为各向异性的液晶,不透明,如果电压以某种图形加在玻璃板上,便产生图象。

110
2、液晶高分子纤维 第一个被发现可以作为高性能纤维材料的合成液晶高分子材料为聚对苯甲酰胺,但由于原料的原因,没有实现工业化,第一个实现工业化的液晶高分子是聚对苯二甲酰对苯二胺(1972年,Kevlar纤维) 液晶纺丝技术:液晶高分子的流变学特性为:高浓度、高粘度和低切变速率下的高取向度,因此采用液晶纺丝,可以解决通常高浓度必然带来高粘度的问题。 例如:当纺丝液的温度为90ºC,聚对苯二甲酰对苯二胺/浓硫酸溶液的浓度可以提高到20%作用,而一般的纺丝液浓度只能达到12%左右,而且液晶高分子本身具有取向性,可以在较低的牵伸力下获得高取向度。

111
LCD – Liquid crystal display

112
Kevlar – PPTA – Poly(p-phenylene terephthalamie)
")
113
Applications of Kevlar

114
第九节 共混高聚物的织态结构 高分子混合物 1、高分子混合物分为三类:高分子-增塑剂混合物(增塑高聚物)、高分子-填充剂混合物(增强高聚物)和高分子-高分子混合物(共混高分子、高分子合金)。 2、高分子共混包括物理共混和化学共混两种;从相容性上分为:各组分分子水平混合成均相体系;各组分自成一相,后者更为重要。

115
2.9.2高分子的相容性 1、 要实现完全混合,必须使混合自由能小于零: ⊿F= ⊿H-T⊿S≤0
由于高分子的分子量很大,混合时,熵的变化很小,而且高分子的混合一般为吸热过程,即焓变为正,因此,要使混合自由能为负是困难的。 2、高分子的相容性不象小分子那么简单。不只是指示相容与不相容,而且还有相容性的好坏。 3、相似相容原则。溶度参数接近,但并不总是有效的。 4、以试验方法来判断高分子的相容性。 以相同溶剂溶解后,再混合,通过分相情况判断其相容性,混合液分相的相体积可以作为相容性的一种量度。但有缺陷,溶液与固体是有差别的。 此外,有浇膜、热压成片、熔融成片等的光洁度和透明度来判断。

116
相容性的表征 透明:相容性好 浑浊:相容性差 直接观察共混物的透光性
TEM (Transmission electron microscopy)透射电镜和SEM (Scanning electron microscopy)扫描电镜观察分散相粒子大小 测量共混物的 Tg - 玻璃化转变温度 (Glass transition temperature)的变化
透射电镜和SEM (Scanning electron microscopy)扫描电镜观察分散相粒子大小. 测量共混物的 Tg - 玻璃化转变温度 (Glass transition temperature)的变化.")
117
ABS- poly(acrylonitrile-co-butadiene-co--styrene)
")
118
共混高聚物聚集态的主要特点 1、共混高分子处于一种准稳定态。热力学不稳定,动力学稳定。但嵌段共聚物形成的非均相体系是热力学稳定的。 2、共混高分子混合物的分散程度取决于组分间的相容性。相容性太差,易于造成宏观相分离,甚至肉眼可见;相容性适中的混合物才具有实用价值,在某些性能上表现出优异的性能,其相分离是微观或亚微观的相分离,在外观上是均匀的,甚至在光学显微镜下也观察不出分相现象,但在电镜下仍可观察到。 完全混溶的高分子,除了少数由于协同作用有实用价值,通常没有实际价值。

119
增容方法 (1) 原位增容 Nylon/PP PS-b-PMMA (2) 加入第三组分 PS/PMMA PS/Nylon PS-g-PEO
Ethylene propylene rubber PE/PP PMMA/PS/PAN PMMA/SAN

120
2、光学性能 大多数非均相的共混物不再具有透明性,如ABS塑料为乳白色,连续相AS透明,分散相丁苯胶也透明。
2.9.5 共混高聚物的聚集态结构对性能的影响 1、根据分相情况将共混高分子分为四类:1)分散相软、连续相硬,如,橡胶增韧塑料;2)分散相硬,连续相软,如热塑性弹性体SBS;3)都软,橡胶共混物;4)均硬,如PE改性PC。 2、光学性能 大多数非均相的共混物不再具有透明性,如ABS塑料为乳白色,连续相AS透明,分散相丁苯胶也透明。 PMMA→MBS →控制两相组成,使两相折光率相近,可以得到透明高抗冲MBS 透明SBS塑料(嵌段共聚物)PB段为连续相,PS分散其中,微区尺寸小到10nm,不影响光线通过.
分散相软、连续相硬,如,橡胶增韧塑料;2)分散相硬,连续相软,如热塑性弹性体SBS;3)都软,橡胶共混物;4)均硬,如PE改性PC。 2、光学性能 大多数非均相的共混物不再具有透明性,如ABS塑料为乳白色,连续相AS透明,分散相丁苯胶也透明。 PMMA→MBS →控制两相组成,使两相折光率相近,可以得到透明高抗冲MBS. 透明SBS塑料(嵌段共聚物)PB段为连续相,PS分散其中,微区尺寸小到10nm,不影响光线通过.")
121
3、热性能 非晶高聚物使用温度上限为Tg,增塑可以提高韧性,但降低Tg,使用温度上限下降,但通过共混,则不会降低Tg,如橡胶增韧塑料可以大幅度提高韧性而又不降低使用温度,高抗冲PS塑料。

Similar presentations












>")








