Download presentation
Presentation is loading. Please wait.

1
第四章 药物定量分析 与分析方法验证 第一节 定量分析样品的前处理方法

2
一、概述 1.结构特征与分析方法之间的关系 ①含卤素有机药物 R-X (F,Cl, Br, I )
卤素与芳环相连 Ar-X 结合牢固,需有机破坏 卤素与脂肪链相连 R-X 结合较不牢固,不需有机破坏

3
有机金属药物 R-Me ,结合牢固,适当处理后测定
②含金属有机药物 含金属有机药物 R-O-Me,有机酸或酚的金属盐/配位化合物; 结合不牢固,可直接测定 有机金属药物 R-Me ,结合牢固,适当处理后测定

4
2、药物分析中常用的分析方法 不经有机破坏的分析方法 经有机破坏的分析方法

5
(一) 不经有机破坏的分析方法: ①直接测定法 凡金属原子不直接与碳原子相连的含金属药物或某些C-M(金属原子直接与碳原子相连)键结合不牢固的有机金属药物,在水溶液中可以电离,可选用本法。
 不经有机破坏的分析方法: ①直接测定法 凡金属原子不直接与碳原子相连的含金属药物或某些C-M(金属原子直接与碳原子相连)键结合不牢固的有机金属药物,在水溶液中可以电离,可选用本法。")
6
例1:富马酸亚铁的含量测定(铈量法,ChP 2000)
终点: 邻二氮菲(+Fe3+) 溶于热稀矿酸 Fe2+ Ce4+ Fe3+ 指示剂:邻二氮菲 (+Fe2+) 红色 淡蓝色 滴定的结果用空白试验校正。 每1ml硫酸铈滴定液(0.1mol/L)相当于16.99mg富马酸亚铁。
 溶于热稀矿酸. Fe2+ Ce4+ Fe3+ 指示剂:邻二氮菲. (+Fe2+) 红色. 淡蓝色. 滴定的结果用空白试验校正。 每1ml硫酸铈滴定液(0.1mol/L)相当于16.99mg富马酸亚铁。")
7
例2、葡萄糖酸锑钠的测定 ChP(2000) 取本品约0.3g,精密称定,置具塞锥形瓶中,加水100ml、盐酸15ml与碘化钾试液10ml,密塞、振摇后,在暗处静置10min,用硫代硫酸钠滴定液(0.1mol/L)滴定,至近终点时,加淀粉指示液,继续滴定至蓝色消失,并将滴定的结果用空白试验校正。每lml的硫代硫酸钠滴定液(0.1mol/L)相当于6.088mg的Sb。
滴定,至近终点时,加淀粉指示液,继续滴定至蓝色消失,并将滴定的结果用空白试验校正。每lml的硫代硫酸钠滴定液(0.1mol/L)相当于6.088mg的Sb。")
8
例3、枸橼酸铁铵的测定 ChP(2000) 取本品约0.5g,精密称定,置具塞锥形瓶中。加水15ml溶解后,加硫酸lml,加热至溶液由暗棕色变成淡黄色,放冷至约15℃,滴加高锰酸钾试液至溶液显粉红色并持续5s,加盐酸15ml与碘化钾试液15ml,密塞,静置3min,加水50ml稀释后,用硫代硫酸钠滴定液(0.1mol/L)滴定至近终点时,加淀粉指示液2ml,继续滴定至蓝色消失,并将滴定的结果用空白试验校正。每lml的硫代硫酸钠滴定液(0.1mol/L)相当于5.585mg的Fe。
滴定至近终点时,加淀粉指示液2ml,继续滴定至蓝色消失,并将滴定的结果用空白试验校正。每lml的硫代硫酸钠滴定液(0.1mol/L)相当于5.585mg的Fe。")
9
(二)经水解后测定法 1.碱水解后测定法 是将含卤素的有机药物溶于适当溶剂(如乙醇)中,加氢氧化钠溶液或硝酸银溶液后,加热回流使其水解,将有机结合的卤素经水解作用转变为无机的卤素离子,然后选用间接银量法进行测定。本法适用于含卤素有机药物结构中卤素原子结合不牢固的药物,如卤素和脂肪碳链相连者。
经水解后测定法 1.碱水解后测定法 是将含卤素的有机药物溶于适当溶剂(如乙醇)中,加氢氧化钠溶液或硝酸银溶液后,加热回流使其水解,将有机结合的卤素经水解作用转变为无机的卤素离子,然后选用间接银量法进行测定。本法适用于含卤素有机药物结构中卤素原子结合不牢固的药物,如卤素和脂肪碳链相连者。")
10
例:三氯叔丁醇的测定 三氯叔丁醇在氢氧化钠溶液中加热回流水解,氯元素全部转变成氯化钠,然后用剩余滴定法,即于水解液中加硝酸酸化,再加入定量过量的硝酸银滴定液,使Cl-生成AgCl沉淀,过量的硝酸银,以Fe3+为指示剂,用硫氰酸铵液回滴定。

11
CCl3-C(CH3)2-OH + 4NaOH NaCl + AgNO3 AgCl↓ + NaNO3 (Ksp=1.56×10–10 )
△ 回流 (CH3)2-CO + 3NaCl + HCOONa + 2H2O NaCl + AgNO3 AgCl↓ + NaNO3 (Ksp=1.56×10–10 ) AgNO3 + NH4SCN AgSCN ↓ + NH4NO3 (Ksp=1.0×10-12 ) Fe SCNˉ Fe (SCN) 2+ (淡棕红色)
2-CO + 3NaCl + HCOONa + 2H2O. NaCl + AgNO3. AgCl↓ + NaNO3. (Ksp=1.56×10–10 ) AgNO3 + NH4SCN. AgSCN ↓ + NH4NO3. (Ksp=1.0×10-12 ) Fe3+ + SCNˉ Fe (SCN) 2+ (淡棕红色)")
12
测定方法 取本品约0.1g,精密称定,加乙醇5ml溶解后,加20%氢氧化钠溶液5ml,加热回流15min,放冷至室温,加水20ml与硝酸5ml,精密加硝酸银滴定液(0.1mol/L) 30ml,再加邻苯二甲酸二丁酯5ml,密塞,强力振摇后,加硫酸铁胺指示液2ml,用硫氰酸胺滴定液(0.1mol/L)滴定,并将滴定的结果用空白试验校正。每1ml硝酸银滴定液(0.1mol/L)相当于6.216mg的C4H7Cl3O·1/2H2O。
 30ml,再加邻苯二甲酸二丁酯5ml,密塞,强力振摇后,加硫酸铁胺指示液2ml,用硫氰酸胺滴定液(0.1mol/L)滴定,并将滴定的结果用空白试验校正。每1ml硝酸银滴定液(0.1mol/L)相当于6.216mg的C4H7Cl3O·1/2H2O。")
13
反应摩尔比为1∶3 T——滴定度,每1ml滴定液相当于被测组分的mg数 c——滴定液浓度,mol/L M——被测物的摩尔质量,g/mol
n——1mol样品消耗滴定液的摩尔数,常用反应摩尔比体现,即反应摩尔比为1∶n

14
2.酸水解后测定法 例 硬脂酸镁的测定 ChP(2000) 取本品约0.1g,精密称定,精密加硫酸滴定液(0.05mol/L) 50ml,煮沸至油滴澄清,继续加热10min,放冷至室温,加甲基橙指示液1~ 2滴,用氢氧化钠滴定液(0.1mol/L)滴定。每1ml硫酸滴定液(0.05mol/L)相当于2.016mg的MgO。
 50ml,煮沸至油滴澄清,继续加热10min,放冷至室温,加甲基橙指示液1~ 2滴,用氢氧化钠滴定液(0.1mol/L)滴定。每1ml硫酸滴定液(0.05mol/L)相当于2.016mg的MgO。")
15
Mg(C17 H35COO)2 + H2SO4 MgSO4 + 2 C17 H35COOH 2NaOH + H2SO4
原理: Mg(C17 H35COO)2 + H2SO4 △ MgSO C17 H35COOH 2NaOH + H2SO4 Na2SO4 + 2H2O
2 + H2SO4. △ MgSO4 + 2 C17 H35COOH. 2NaOH + H2SO4. Na2SO4 + 2H2O.")
16
某些含汞有机药物,由于C—Hg键不牢固,在水溶液(冰浴)中滴加硫酸使其水解,将有机汞定量转化成硫酸汞,再用硫氰酸铵滴定液滴定。
中滴加硫酸使其水解,将有机汞定量转化成硫酸汞,再用硫氰酸铵滴定液滴定。")
17
例 汞撒利的测定 取本品约0.5g,精密称定,加蒸馏水10ml,置冰浴中,滴加硫酸10ml,随滴随搅拌,至产生的沉淀复溶解,再加蒸馏水50ml,硫酸铁铵指示液2ml,用硫氰酸铵滴定液(0.1mol/L)滴定。每1ml硫氰酸铵滴定液(0.1mol/L)相当于25.29mg的汞撒利。
滴定。每1ml硫氰酸铵滴定液(0.1mol/L)相当于25.29mg的汞撒利。")
18
3、经还原分解后测定法 ①碱性还原后测定 卤素结合于芳环上时,由于分子中碘的结合较牢固,需在碱性溶液中加还原剂(如锌粉)回流,使碳-碘键断裂,形成无机碘化物后测定。 ②酸性还原后测定法 两法均是将含卤素有机药物在碱性或酸性下,加还原剂(如锌粉)加热回流,药物产生还原裂解反应,使有机结合的卤素转变为无机的卤素离子,然后采用银量法(Fajans法)测定。
回流,使碳-碘键断裂,形成无机碘化物后测定。 ②酸性还原后测定法 两法均是将含卤素有机药物在碱性或酸性下,加还原剂(如锌粉)加热回流,药物产生还原裂解反应,使有机结合的卤素转变为无机的卤素离子,然后采用银量法(Fajans法)测定。")
19
碱性还原后测定法例 泛影酸的测定 ChP (2000)
取本品约0.4g,精密称定,加氢氧化钠溶液30ml与锌粉1.0g,加热回流30min,放冷,冷凝管用少量水洗涤,滤过,烧瓶与滤器用水洗涤3次,每次15ml,洗液与滤液合并,加冰醋酸5ml与曙红钠指示液5滴,用硝酸银滴定液(0.1mol/L)滴定。每1ml硝酸银滴定液(0.1mol/L)相当于20.46mg的C11H9 I 3N2O4。
滴定。每1ml硝酸银滴定液(0.1mol/L)相当于20.46mg的C11H9 I 3N2O4。")
20
指示剂:曙红钠 终点:黄→红

21
ChP(2000)收载的胆影酸、碘番酸、胆影葡胺、泛影葡胺、碘他拉酸等均采用同法测定。
USP (24)、BP(1998)
、BP(1998)")
22
经还原--汞齐化后测定法(含汞有机药物的含量测定)
本法系在酸性或碱性溶液中,加锌粉、加热回流,将药物中有机结合的汞还原析出金属汞,并与过量的锌生成锌汞齐。将锌汞齐溶于硝酸后,选用适当的方法测定汞的含量,并换算成含汞有机药物的含量。

23
如醋酸苯汞的含量测定 取本品约0.5g,精密称定,置100ml烧瓶中,加水15ml,甲酸5ml与锌粉1g,附回流冷凝器,煮沸30min。放冷,滤过,滤纸和锌汞齐用蒸馏水洗涤至洗液对石蕊试纸不显酸性反应。将锌汞齐溶解在稀硝酸(1∶2)40ml中,置蒸气浴上加热3min,加尿素0.5g和足够的高锰酸钾试液至显桃红色。冷却后,加过氧化氢溶液脱色,加硫酸铁铵指示剂lml,用硫氰酸铵滴定液(0.1mol/L)滴定,即得。每1ml硫氰酸铵液(0.1mol/1)相当于16.84mg的C8H8O2Hg。
40ml中,置蒸气浴上加热3min,加尿素0.5g和足够的高锰酸钾试液至显桃红色。冷却后,加过氧化氢溶液脱色,加硫酸铁铵指示剂lml,用硫氰酸铵滴定液(0.1mol/L)滴定,即得。每1ml硫氰酸铵液(0.1mol/1)相当于16.84mg的C8H8O2Hg。")
24
三、经有机破坏的分析方法 金属原子、卤素等与C结合牢固者,必须有机破坏 湿法破坏 药物分析中常用的有机破坏的方法有 H2SO4-HNO3法 (一)湿法破坏 H2SO4-HClO4法 H2SO4-硫酸盐法 H2SO4-H2O2,HNO3-KMnO4法等 干法破坏 ----凯氏定氮法

25
(一)湿法破坏 1、硝酸-高氯酸法: 适于生物样品的破坏,如血、尿、组织等。。反应剧烈、切勿蒸干、以防爆炸但对含氮杂环破坏不完全,应用干法灼烧。无机金属离子一般为高价态。 2、硝酸-硫酸法: 适于大多数有机药物,如染料、中间体、药物等。但不用于碱土金属有机药物(用硝酸-高氯酸法)
湿法破坏 1、硝酸-高氯酸法: 适于生物样品的破坏,如血、尿、组织等。。反应剧烈、切勿蒸干、以防爆炸但对含氮杂环破坏不完全,应用干法灼烧。无机金属离子一般为高价态。 2、硝酸-硫酸法: 适于大多数有机药物,如染料、中间体、药物等。但不用于碱土金属有机药物(用硝酸-高氯酸法)")
26
3、H2SO4—硫酸盐法 用于含砷或锑有机药物破坏。所用硫酸盐为硫酸钾或硫酸钠,目的是为了提高硫酸沸点,使其破坏完全。无机金属离子为低价态。 4、其他湿法 尚有硝酸-硫酸-高氯酸法、硫酸-过氧化氢法、硫酸-高锰酸钾法。 湿法破坏一般用硅玻璃或硼玻璃制的凯氏烧瓶。进行空白试验校正。含金属元素10-100μ g范围内,取样10g;生物样品:血样10-15ml,尿样50ml。

27
注意事项: 1. 湿法破坏所用的仪器,一般为硅玻璃或硼玻璃制成的凯氏烧瓶; 2
注意事项: 1.湿法破坏所用的仪器,一般为硅玻璃或硼玻璃制成的凯氏烧瓶; 2.所用试剂及蒸馏水均不应含有被测金属离子或干扰测定的其他金属离子等组分; 3.由于整个操作过程所用矿酸量数倍于样品,所以必须按相同条件进行空白试验校正; 4.操作时应在通风橱内进行。

28
样品的取用量,应视被测含金属有机药物中所含金属元素的量和破坏后所用测定方法而定。一般来说,含金属元素量在10μg一100μg范围内时,取样量为10g;如果测定方法灵敏度较高,取样量可相应减少。对于生物样品,一般血样10ml一15ml或尿样50ml。

29
凯氏定氮法: 原理:在催化作用下,用浓硫酸将有机物分解破坏,使有机分子中的N转变为(NH4)2SO4,加碱碱化后使(NH4)2SO4分解,将滴出的NH3蒸馏在H3BO3溶液中,用H2SO4标准液滴定至终点。根据消耗的量计算出有机物中N的量。 四步骤:消解、蒸馏、吸收、滴定。 试剂:浓硫酸:氧化剂、碳化剂; 硫酸钾:提高H2SO4的沸点; 硫酸铜:催化剂,使之更完全、更彻底。 注意事项:从消解开始做空白试验。

32
(二)干法破坏:系将有机物灼烧灰化以达分解目的。 适用于含卤素、S、P等有机药物分析的前处理,也用于某些药物中Se及砷盐的检查
1. 高温炽灼法 加无水Na2CO3、硝酸镁、氢氧化钙或ZnO以助灰化,先小火加热,使样品完全炭化,再高温炉中灼烧,使其完全灰化。 适用于:湿法不易破坏的有机物以及某些不能用硫酸进行破坏的有机药物; 不适用于:含易挥发性金属(如汞、砷等)有机破坏。
有机破坏。")
33
应用本法时要注意以下几个问题: ①加热或灼烧时,应控制温度在420℃以下,以防止某些被测金属花合物的挥发。 ②经本法破坏后,所得灰分往往不易溶解,但此时切勿弃去。 ③灰化完全与否,直接彭响侧定结果的准确性。

34
(三)氧瓶燃烧法 ①定义:氧瓶燃烧法(oxygen flask combustion method)系将有机药物放入充满氧气的密闭烧瓶中进行燃烧,将产生的欲测物吸收于吸收液中,再用适宜方法测定。适于卤素有机药物或含硫、氮、硒等其他元素的有机药物。其吸收液多是水或水与氢氧化钠的混合液。
氧瓶燃烧法 ①定义:氧瓶燃烧法(oxygen flask combustion method)系将有机药物放入充满氧气的密闭烧瓶中进行燃烧,将产生的欲测物吸收于吸收液中,再用适宜方法测定。适于卤素有机药物或含硫、氮、硒等其他元素的有机药物。其吸收液多是水或水与氢氧化钠的混合液。")
35
②称样用材料及称样 A. 固体样品 无灰滤纸 B. 液体样品 纸袋 C. 软膏类样品:将适量样品置不含被测成分的蜡油纸中包裹严密,外层再用无灰滤纸包裹 ③氧气

36
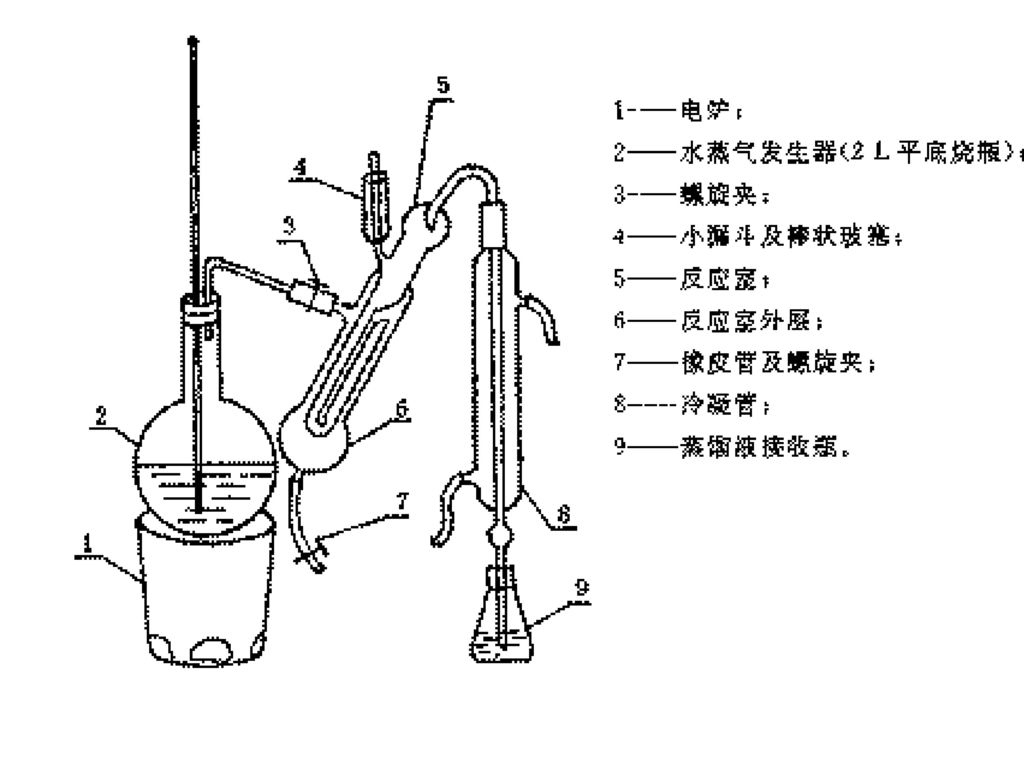
氧瓶燃烧法装置与样品包装操作图

37
④吸收液的选择 吸收液可使样品经燃烧分解所产生的各种价态的卤素,定量地被吸收并使其转变为一定的便于测定的价态,以适应所选择的分析方法。用于X、S、Se等的鉴别、检查、含量测定时多数是H2O或H2O-NaOH;少数为H2O-NaOH-H2O2。 氟 水 氯 NaOH溶液 溴 H2O2-NaOH NaOH-硫酸肼饱和溶液 碘 NaOH-硫酸肼饱和溶液 硫 NaOH或H2O2 硒 硝酸溶液

38
⑤注意事项 A、根据被燃烧分解的样品量选用适宜大小的燃烧瓶。 ChP(2000)规定的燃烧瓶体积为500、1000或2000ml三种,采用常量或半微量分析
规定的燃烧瓶体积为500、1000或2000ml三种,采用常量或半微量分析")
39
待分解样品量 燃烧瓶的体积 3~5mg(微量分析) 150 ~ 250ml 20 ~ 30mg(半微量分析) 300 ~ 500ml 50 ~ 60mg 1000ml 0.6 ~ 0.7g或更多 2000ml或特殊结构的燃烧瓶

40
正确选用燃烧瓶的目的在于: 样品能在足够的氧气中燃烧分解完全; 有利于将燃烧分解产物较快地吸收到吸收液中; 防止爆炸的可能性。

41
B、测定含氟有机药物时,用石英制燃烧瓶 C、铂丝燃烧时起催化作用 D、应同时做空白试验 E、燃烧时要注意防爆 F、燃烧要完全 G、燃烧产生的烟雾完全被吸收

42
例 碘苯酯的含量测定 ChP(2000) 原理 碘苯酯系有机碘化物,用氧瓶燃烧分解,转变为碘化物,继而氧化为游离的碘,并被定量地吸收于吸收液中,和氢氧化钠反应,生成碘化物与碘酸盐,加入溴-醋酸溶液,使全部转变为碘酸盐,过量的溴以甲酸及通空气去除。加入碘化钾,使与碘酸盐反应析出游离碘,用硫代硫酸钠液滴定,碘与淀粉结合所显的蓝色消失即为终点。

43
应用示例:碘苯酯的测定

44
1molNaIO→1/3molNaIO3+2/3molNaI
滴定度的计算 放大反应 2mol碘苯酯→1molI2 1molI2→1molNaIO+1molNaI 1molNaIO→1/3molNaIO3+2/3molNaI molNaI+Br2→ molNaIO3 即2mol碘苯酯→2molNaIO3

45
2mol NaIO3+10I-→6molI2 即2mol碘苯酯→1molI2 →6molI2→12molI→12molNa2S2O3 反应摩尔比为1∶6

46
第二节 定量分析方法特点 一、容量分析法 (一)容量分析法的特点 几个概念:容量分析法、滴定终点、滴定误差、等当点 操作简单、快速;
第二节 定量分析方法特点 一、容量分析法 (一)容量分析法的特点 几个概念:容量分析法、滴定终点、滴定误差、等当点 操作简单、快速; 比较准确(相对误差<0.2%); 仪器普通易得 常用于测定高含量或中含量组分,原料药含量测定首选
容量分析法的特点. 几个概念:容量分析法、滴定终点、滴定误差、等当点. 操作简单、快速; 比较准确(相对误差<0.2%); 仪器普通易得. 常用于测定高含量或中含量组分,原料药含量测定首选.")
47
容量分析法用原料药精制品(含量>99. 5%)或对照品考察方法的精密度,相对标准差一般应不大于0
容量分析法用原料药精制品(含量>99.5%)或对照品考察方法的精密度,相对标准差一般应不大于0.2%;进行回收率试验。回收率一般在99.7~100.3%之间。
或对照品考察方法的精密度,相对标准差一般应不大于0.2%;进行回收率试验。回收率一般在99.7~100.3%之间。")
48
二、光谱分析法 光谱 光谱分析法 分光光度法 分类 紫外-可见分光光度法 红外分光光度法 原子吸收分光光度法 荧光分光光度法 火焰光度法

49
仪器价格低廉,操作简单,易普及,应用广范 2. 朗伯-比耳定律 A = ECL 吸收系数:摩尔吸收系数ε —— 研究分子结构
二、光谱分析法 (一)紫外-可见分光光度法 吸收光谱范围:紫外:200~400nm 可见:400~760nm 灵敏度高,可达10-4g/ml ~ 10-7g/ml 1. 特点 准确度高,RSD(%)为2% ~5% 仪器价格低廉,操作简单,易普及,应用广范 2. 朗伯-比耳定律 A = ECL 吸收系数:摩尔吸收系数ε —— 研究分子结构 百分吸收系数E —— 含量测定
紫外-可见分光光度法. 吸收光谱范围:紫外:200~400nm. 可见:400~760nm. 灵敏度高,可达10-4g/ml ~ 10-7g/ml. 1. 特点 准确度高,RSD(%)为2% ~5% 仪器价格低廉,操作简单,易普及,应用广范. 2. 朗伯-比耳定律 A = ECL. 吸收系数:摩尔吸收系数ε —— 研究分子结构. 百分吸收系数E —— 含量测定.")
50
3.仪器的校正和检定 波长的校正:汞灯中的几根较强的谱线或用仪器自身 所带的氘灯的特定谱线为参照进行校正 吸收度准确性的检定:重铬酸钾的硫酸溶液,规定波 长处测定E ,应符合P51表1中规定 杂散光的检查:一定浓度的碘化钠和亚硝酸钠溶液, 规定波长处测定透光率,应符合P51表2 中规定

51
含杂原子的有机溶剂通常具有很强的末端吸收 它们的使用范围均不能小于截止使用波长 石英吸收池、空气为空白 λ(nm) 溶剂+吸收池A
4. 对溶剂的要求: 含杂原子的有机溶剂通常具有很强的末端吸收 它们的使用范围均不能小于截止使用波长 石英吸收池、空气为空白 λ(nm) 溶剂+吸收池A 220~ ≤0.40 241~ ≤ 0.20 251~ ≤ 0.10 300以上 ≤ 0.05
 溶剂+吸收池A. 220~240 ≤ ~250 ≤ ~300 ≤ 以上 ≤")
52
5. 测定方法 要求供试品溶液的A应在0.3 ~0.7 1)对照品比较法:A供/A对=C供/C对 原料药: 制剂:
对照品比较法:A供/A对=C供/C对 原料药: 制剂:")
53
2)吸收系数法: 注意: 应>100 应注意仪器的校正和检定: 作为计算浓度的因数进行定量的方法,不是任何情况下都适用的,特别时单色光不纯的情况下,A会随仪器不同而变化,误差较大 但若认定一台仪器,固定其工作状态和测定条件,则C和A间的关系在多数情况下仍符合Lambert-Beer定律

54
3)计算分光光度法: 多种,具体按每种药物规定的方法进行 如:VA的三点校正法 4)比色法 加入显色剂后,按照对照品比较法测定 影响显色因素很多,故需注意平行操作 空白溶剂:溶剂+显色剂,同法处理
计算分光光度法: 多种,具体按每种药物规定的方法进行 如:VA的三点校正法 4)比色法 加入显色剂后,按照对照品比较法测定 影响显色因素很多,故需注意平行操作 空白溶剂:溶剂+显色剂,同法处理")
55
(二)荧光分析法 干扰因素多,需做空白实验 在低浓度进行测定,防止自熄灭作用 1. 特点 用基准溶液代替对照液校正仪器灵敏度
灵敏度高,可达10-10g/ml ~ 10-12g/ml 在低浓度进行测定,防止自熄灭作用 1. 特点 用基准溶液代替对照液校正仪器灵敏度 制备荧光衍生物,可提高灵敏度和选择性 干扰因素多,需做空白实验 样品用量少,操作简单,应用范围较广

56
2.含量测定 计算: 原理:当激发光强度、波长、所用溶剂及温度等条件固定时,物质在一定浓度范围内,其荧光强度R与该物质浓度C成正比
方法:对照品比较法 计算:

57
根据混合物中各组分的色谱行为差异,先行分离后再在线或离线对各组分逐一进行分析的方法,是分离分析混合物的最有力手段。
三、色谱分析法 根据混合物中各组分的色谱行为差异,先行分离后再在线或离线对各组分逐一进行分析的方法,是分离分析混合物的最有力手段。 按分离原理:吸附;分配;离子交换;排阻色谱 分类 按分离方法:PC;TLC;柱色谱;GC;HPLC 特点:高灵敏度、 高效能、高速度、应用广泛

58
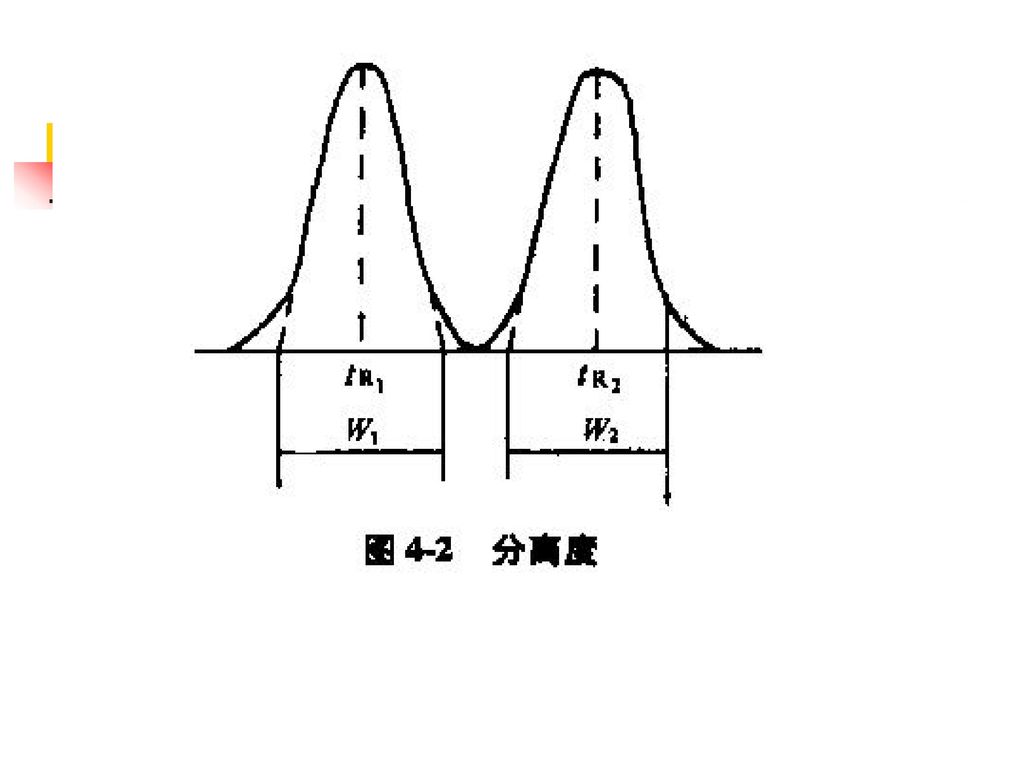
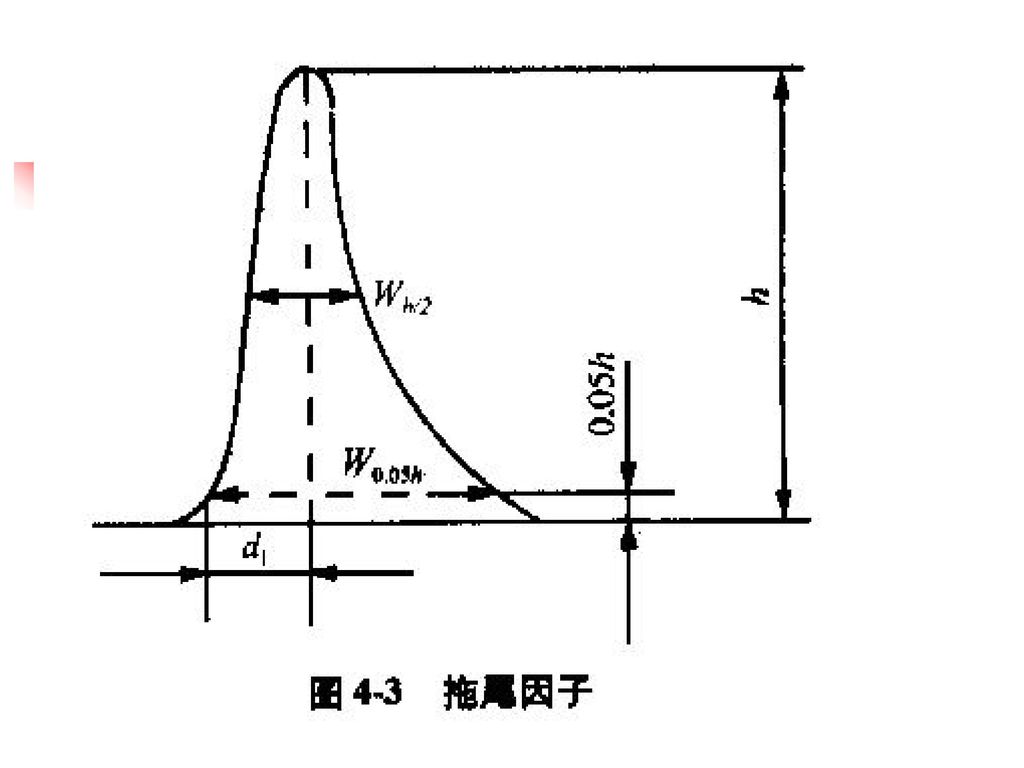
(一)HPLC法 1. 对仪器一般要求 固定相种类、流动相组分、检测器类型不得任意改变 其他均可适当改变 色谱图20分钟内记录完毕 2. 系统适用性试验 色谱柱的理论板数:n = 5.54(tR /Wh/2)2 分离度:R = 2(tR1 – tR2)/(W1 + W2); 应大于1.5 重复性:对照液连续进样5次,峰面积RSD≤2.0% 拖尾因子:T = W0.05h/2d1 应在0.9 ~ 1.05
/(W1 + W2); 应大于1.5. 重复性:对照液连续进样5次,峰面积RSD≤2.0% 拖尾因子:T = W0.05h/2d1 应在0.9 ~")
59
1)内标法加校正因子测定供试品中主成分含量
测定方法 1)内标法加校正因子测定供试品中主成分含量 峰面积 含量 对照液(R): AR CR AS/CS f= ———— AR /CR 内标液(S): AS CS A’S / C’S f= ———— Ax /Cx 供试液(X): AX CX Ax Cx=f· ———— A’S / C’S
内标法加校正因子测定供试品中主成分含量. 峰面积 含量. 对照液(R): AR CR. AS/CS. f= ———— AR /CR. 内标液(S): AS CS. A’S / C’S. f= ———— Ax /Cx. 供试液(X): AX CX. Ax. Cx=f· ———— A’S / C’S.")
60
2)外标法测定供试品中主成分含量 对照液(R): AR CR CX CR ——=—— AX AR 供试液(X): AX CX CR· AX
要求:进样量准确、操作条件稳定

61
(二)GC法 1. 对GC仪器一般要求 载 气:氮气 色谱柱:填充柱或毛细管柱 填充柱:内径2~4mm,长1~10m,内装吸附剂 高分子多孔小球或涂渍固定液的载体 毛细管柱:内径0.2~0.5mm,长10~100m,一般 为空心柱,内壁或载体经涂渍或交联 固定液 检测器:氢火焰离子化检测器 色谱图:30分钟内记录完毕

62
2. 色谱适用性实验:同HPLC 3.测定法:同HPLC

63
验证内容:准确度、精密度、专属性、检测限、定量限、线性、范围和耐用性
第三节 药品分析方法验证 目的:证明采用的方法适合于相应检测要求。 起草药品质量标准 生产工艺变更 制剂组分改变 原分析方法修订 均需对分析方法进行验证 验证内容:准确度、精密度、专属性、检测限、定量限、线性、范围和耐用性

64
一、准确度 是指用该方法测定结果与真实值接近的程度,用回收率(%)表示 (一)含量测定方法的准确度 1. 原料药:可用已知纯度对照品或供试品进行测定;或与已知准确度的另一方法测定的结果进行比较
表示 (一)含量测定方法的准确度 1. 原料药:可用已知纯度对照品或供试品进行测定;或与已知准确度的另一方法测定的结果进行比较")
65
2. 制剂:考察其他组分和辅料对回收率的影响 ①用含已知量被测物的制剂各组分混合物(包括制剂辅料)进行测定,回收率计算同原料药 ②向制剂中加入已知量的被测物进行测定 ③与已知准确度的另一方法测定的结果进行比较 (二)具体做法:测定高、中、低三个浓度,n=3, 共9个数据来评价回收率;用UV和HPLC法时,一般回收率可达98%~102%;容量法可达99.7%~100.3%
具体做法:测定高、中、低三个浓度,n=3, 共9个数据来评价回收率;用UV和HPLC法时,一般回收率可达98%~102%;容量法可达99.7%~100.3%")
66
二、精密度 是指在规定的测试条件下,同一个均匀样品,经多次测定所得结果之间的接近程度。 (一)精密度表示方法
1. 偏差(deviation , d) ★ d = 测得值-平均值=Xi-X ★ 相对偏差(RD)=d / X ×100% 实验中:两份平行操作 RD=(A-B)/(A+B)×100% ★最大相对偏差(允许差) 容量分析法0.3%,仪器分析法3%
 ★ d = 测得值-平均值=Xi-X. ★ 相对偏差(RD)=d / X ×100% 实验中:两份平行操作. RD=(A-B)/(A+B)×100% ★最大相对偏差(允许差) 容量分析法0.3%,仪器分析法3%")
67
2. 标准偏差(standard deviation, SD或S) S =[(∑Xi – X)2/(n-1)]1/2
3. 相对标准偏差(relative standard deviation, RSD) ,也称变异系数(coefficient of variation,CV) RSD=标准偏差/平均值×100%=S/X ×100% 容量法分析原料药时:平行试验5个样本,试验数据的RSD一般应不大于0.2%;UV法用适当浓度的精制品进行测定,其RSD一般不大于1%(n=3~5);HPLC法要求RSD小于2%(n=3~5)。
![2. 标准偏差(standard deviation, SD或S) S =[(∑Xi – X)2/(n-1)]1/2](http://slidesplayer.com/slide/11177926/60/images/67/2.+%E6%A0%87%E5%87%86%E5%81%8F%E5%B7%AE%EF%BC%88standard+deviation%2C+SD%E6%88%96S%EF%BC%89+S+%3D%5B%28%E2%88%91Xi+%E2%80%93+X%292%2F%28n-1%29%5D1%2F2.jpg "3. 相对标准偏差(relative standard deviation, RSD) ,也称变异系数(coefficient of variation,CV) RSD=标准偏差/平均值×100%=S/X ×100% 容量法分析原料药时:平行试验5个样本,试验数据的RSD一般应不大于0.2%;UV法用适当浓度的精制品进行测定,其RSD一般不大于1%(n=3~5);HPLC法要求RSD小于2%(n=3~5)。")
68
(二)验证内容 1. 重复性 在相同条件下,由一个分析人员测定所得结果的精密度.
1. 重复性 在相同条件下,由一个分析人员测定所得结果的精密度. 2. 中间精密度 在同一个实验室,不同时间由不同分析 人员用不同设备测定结果的精密度。 3. 重现性 在不同实验室由不同分析人员测定结果的精 密度。 (三)数据要求 均应报告标准偏差、相对标准偏差和可信限
数据要求. 均应报告标准偏差、相对标准偏差和可信限.")
69
三、专属性 指在其他成分(如杂质、降解产物、辅料等)可能存在下,采用的方法能准确测定出被测物的特性 鉴别、杂质检查、含量测定方法,均应考察其专属性 (一)鉴别反应 应能与其它共存的物质或相似化合物区分,不含被测组分的样品均应呈现负反应
鉴别反应. 应能与其它共存的物质或相似化合物区分,不含被测组分的样品均应呈现负反应.")
70
图中应注明各组分的位置,色谱法中分离度应符合要求。 在能获得杂质的情况下,可加到试样中,考察对结果的干扰。
(二)含量测定和杂质测定 色谱法和其他分离法,应附代表性的图谱,以说明专属性。 图中应注明各组分的位置,色谱法中分离度应符合要求。 在能获得杂质的情况下,可加到试样中,考察对结果的干扰。 在杂质和降解物不能获得的情况下,可用以验证方法和药典方法进行对照;也可用对试样加速破坏的方法,比较两种方法。
含量测定和杂质测定. 色谱法和其他分离法,应附代表性的图谱,以说明专属性。 图中应注明各组分的位置,色谱法中分离度应符合要求。 在能获得杂质的情况下,可加到试样中,考察对结果的干扰。 在杂质和降解物不能获得的情况下,可用以验证方法和药典方法进行对照;也可用对试样加速破坏的方法,比较两种方法。")
71
四、检测限(limit of detection,LOD) 是指试样中被测物能被检测出的最低浓度或量,是限度检验指标。它无需测定,只要指出高于或低于该规定的浓度或量即可。
1.非仪器分析目视法 用含已知浓度被测物的试样进行分析,目视确定能被可靠地检测出的被测物的最低浓度或量,常用于显色鉴别法,TLC 2.仪器分析时的测定法 当用紫外分光光度法和荧光分析法时:可做多次空白试验,求得其背景响应值的标准偏差,再将3倍空白标准差作为检测限的估计算。 (2) 当用GC法和HPLC法时,一般以S/N=2或3时的相应浓度来确定检测限。 3. 数据要求:应附测试图谱,说明测试过程和检测限结果。
 当用GC法和HPLC法时,一般以S/N=2或3时的相应浓度来确定检测限。 3. 数据要求:应附测试图谱,说明测试过程和检测限结果。")
72
五、定 量 限 1、定义:定量限(limit of quantitation ,LOQ)是指样品中被测物能被定量测定的最低量,其测定结果应具一定准确度和精密度。 2、确定方法:常用信噪比法确定定量限。杂质和降解产物进行定量测定时,要求LOQ常用信噪比法确定定量限,一般以S/N=10时相应的浓度进行测定。

73
六、线 性 1、定义:线性系指在设计的范围内,测试结果与试样中被测物浓度直接呈正比关系的程度。
六、线 性 1、定义:线性系指在设计的范围内,测试结果与试样中被测物浓度直接呈正比关系的程度。 2、条件:回归方程的相关系数 ( r ) 越接近于 1 表明线性关系越好。 3、示例:在UV中,浓度点n=5,用浓度C对吸收度A作线性回归,得一直线方程,方程截距应近于零,相关系数应大于0.9999。在HPLC中,浓度点n=5~7,用浓度C对峰高h或峰面积A或被测物与内标物的响应值之比进行回归处理,建立回归方程,截距应趋于零,相关系数大于0.9999。 4、数据要求: 应列出回归方程、相关系数和线性图。
 越接近于 1 表明线性关系越好。 3、示例:在UV中,浓度点n=5,用浓度C对吸收度A作线性回归,得一直线方程,方程截距应近于零,相关系数应大于0.9999。在HPLC中,浓度点n=5~7,用浓度C对峰高h或峰面积A或被测物与内标物的响应值之比进行回归处理,建立回归方程,截距应趋于零,相关系数大于0.9999。 4、数据要求: 应列出回归方程、相关系数和线性图。")
74
七、范 围 1、定义:范围系指能达到一定精密度、准确度和线性、测试方法适用的高低限浓度或量的区别。
七、范 围 1、定义:范围系指能达到一定精密度、准确度和线性、测试方法适用的高低限浓度或量的区别。 2、 范围应根据分析方法的具体应用和线性、准确度、精密度结果和要求确定。 3、如原料药和制剂含量测定:范围应为测试浓度的80%~120%;制剂含量均匀度检查:范围应为测试浓度的70%~130%; 4、根据剂型特点,如气雾剂、喷雾剂可适当放宽,溶出度或释放度中的溶出量测定,范围应为限度的± 20%。

75
八、耐 用 性 1、定义: 耐用性系指在测定条件下有小的变动时,测定结果不受影响的承受程度,为常规检验提供依据。
八、耐 用 性 1、定义: 耐用性系指在测定条件下有小的变动时,测定结果不受影响的承受程度,为常规检验提供依据。 2、开始研究分析方法时,就应考虑其耐用性,如果测试条件要求苛刻,则应在方法中写明。 3、验证一种分析方法,分为三种情况: (1)鉴别试验:对专属性、耐用性有要求,其他无; (2)原料药中杂质或制剂中的降解产物分为两种: a、用于定量:除检测限不要求外,其他均要求; b、用于限度检查:对专属性、检测限、耐用性有要求; (3)用于原料药中主成分或制剂中有效组分含量测定及溶出度测定:除了检测限和定量限外,其他六项均要求。
鉴别试验:对专属性、耐用性有要求,其他无; (2)原料药中杂质或制剂中的降解产物分为两种: a、用于定量:除检测限不要求外,其他均要求; b、用于限度检查:对专属性、检测限、耐用性有要求; (3)用于原料药中主成分或制剂中有效组分含量测定及溶出度测定:除了检测限和定量限外,其他六项均要求。")
76
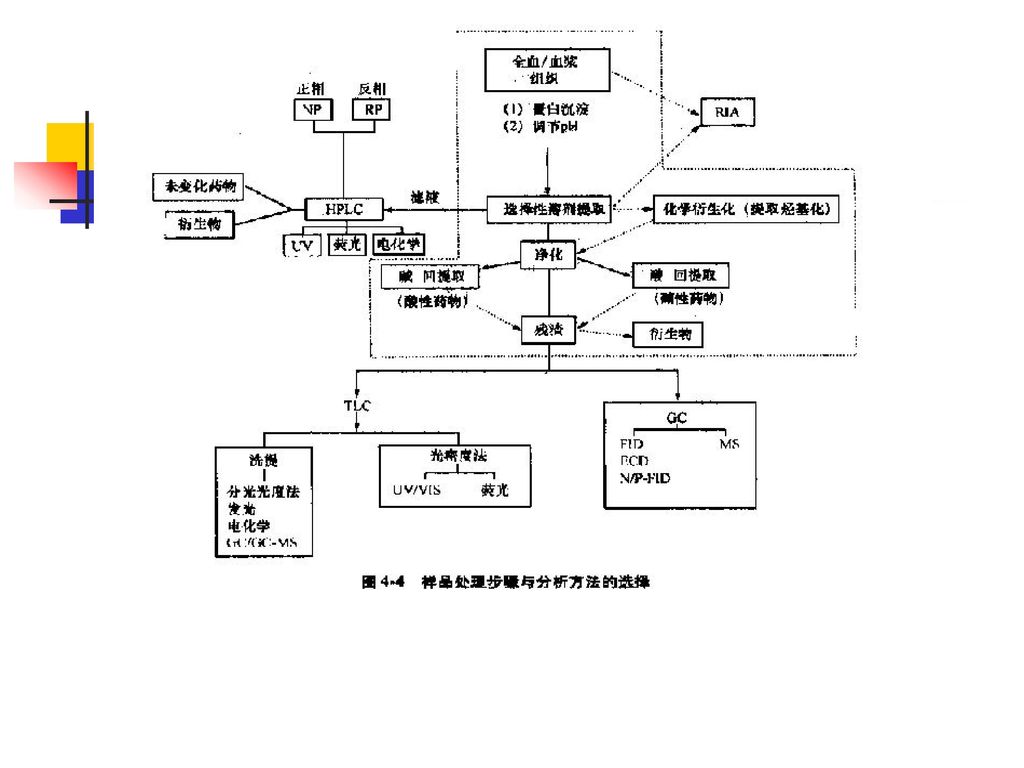
第四节 生物样品分析方法的基本要求 一、常用样品的种类、采集和贮藏 二、生物样品分析前处理技术 三、定量分析方法验证 返 回

77
一、常用样品的种类、采集和贮藏 作用:1、对药物内在质量的评估; 2、给药剂量个体化。
生物样品包括各种体液和组织,但实际上最常用的是比较容易得到的血液(血浆、血清、全血)、尿液、唾液。 任务:A、体内药物及代谢物的测定; B、为TDM提供data and information; C、内源性活性物质(多为酸性物质); D、分析方法的建立; E、分析技术的改进; F、方法学的研究 。 作用:1、对药物内在质量的评估; 2、给药剂量个体化。
、尿液、唾液。 任务:A、体内药物及代谢物的测定; B、为TDM提供data and information; C、内源性活性物质(多为酸性物质); D、分析方法的建立; E、分析技术的改进; F、方法学的研究 。 作用:1、对药物内在质量的评估; 2、给药剂量个体化。")
78
特点:1、样本、对象:体液、血液、组织药物浓度或活度极低,大多数样品需分离纯化;
2、介质复杂:结合物(与蛋白质结合)、缀合物(与硫酸、葡萄糖、醛酸成酯); 3、样品少,不可重新获得,要求一次成功; 样品采集原则: A、浓度与治疗作用有相关性; B、易于获得; C、易处理,适于分析; D、不同目的与要求进行选择。
、缀合物(与硫酸、葡萄糖、醛酸成酯); 3、样品少,不可重新获得,要求一次成功; 样品采集原则: A、浓度与治疗作用有相关性; B、易于获得; C、易处理,适于分析; D、不同目的与要求进行选择。")
79
(一) 血样 1、血药浓度的定义:血中药物浓度通常是指测定血浆或血清中的药物浓度,而不是指含有血细胞的全血中的药物浓度。 2、血样的采集:动物实验时,可直接从动脉或心脏取血.对于病人,通常采取静脉血,有时根据血药浓度和分析方法灵敏度,也可用毛细管采血。由采集的血液制取血浆或血清。 3、血样的保存:采血血样后,应及时分离血浆和血清,并最好立即进行分析。如不能立即测定时,应妥善储存,应注意采血后及时分离出血浆或血清再进行储存。 4、血清(serum),血浆(plasma),全血(whole blood) 测定药物浓度指测定血浆或血清中的药物浓度 采集→制备血浆或血清 贮藏:短期4℃、长期-20 ℃
,血浆(plasma),全血(whole blood) 测定药物浓度指测定血浆或血清中的药物浓度. 采集→制备血浆或血清. 贮藏:短期4℃、长期-20 ℃")
80
(二) 唾液 1、唾液的定义:由腮腺、颌下腺、舌下腺和口腔黏膜内许多散在的小腺体分泌液混合组成的,平时所说的唾液就是指此混合液。 2、唾液的前处理: 唾液的采集应尽可能在刺激少的安静状态下进行,一般淑口后15min收集,采集后应立即测量其除去泡沫部分的体积。放置后分为泡沫部分、透明部分及乳白色沉淀部分三层。分层后,以3000r/min离心分离10min,取上清液作为药物浓度测定的样品。 3、唾液的保存: 唾液在保存过程中,会放出二氧化碳而使PH值升高,因此,需要测定唾液的pH值时,应在取样当时为好,冷藏保存唾液时,解冻后有必要充分搅匀后再用,不然测定结果会产生误差。 采集:自然分泌 贮藏: 4℃以下

81
(三) 尿液 1、尿液主要成分是水、含氮化合物(其中大部分是尿素)及盐类 2、尿液的用途:主要用于药物剂量回收研究、尿清除率、生物利用度的研究,推断患者是否违反医嘱用药;同时根据药物剂量回收研究可以预测药物的代谢过程及测定药物的代谢类型(代谢速率,MR)等。 3、尿液类型:采集的尿是自然排尿。测定尿中药物浓度时应采用时间尿。 4、尿液的保存:采集的尿样应立即测定,若收集24h的尿液不能立即测定时,应加入防腐剂(1%甲苯或氯仿饱和)置冰箱中保存。保存时间为24h~36h,长时间保存时,应冰冻。
置冰箱中保存。保存时间为24h~36h,长时间保存时,应冰冻。")
82
二、生物样品分析前处理技术 为什么要对生物样品进行前处理? 蛋白质的去除方法? 常用的分离、纯化方法? 被测组分的浓集方法?

83
主要考虑问题:生物样品的前处理涉及很多方面,但主要考虑生物样品的种类,被测定药物的性质和测定方法三个方面的问题。
采取前处理方法的依据:样品的分离、纯化技术应该根据生物样品的类型,根据被测定药物的结构、理化性质及药理性质、存在形式、浓度范围等,采取响应的前处理方法。 不同方法不同结果:样品于测定前是否需要纯化以及纯化到什么程度均与其后采用的测定方法的不同而不同。

84
(一) 去除蛋白质 1、加入与水相混溶的有机溶剂:如乙腈、甲醇、乙醇、丙酮、四氢呋喃等 2、加入中性盐:如饱和硫酸铵、硫酸钠、镁盐、磷酸盐及枸橼酸盐等 3、 加入强酸:如10%三氯醋酸、6%高氯酸、硫酸-钨酸混合液、5%偏磷酸等 4、加入含锌盐及铜盐的沉淀剂:CuSO4-Na2WO4、ZnSO4-NaOH等 5、酶解法:枯草菌溶素

85
(二) 缀合物的水解 酸水解/酶水解 尿中药物多数呈缀合状态.无论是直接测定或萃取分离之前,都需要将缀合物中的药物释出.有些药物较温和条件即可使药物游离,有些则需较剧烈的方法,常用酸水解和酶水解的方法 (三) 分离、纯化与浓集 提取法是应用最多的分离、纯化方法: 1、液-液提取法 2、液-固提取法 浓集的方法主要有: 1、是在末次提取时加入的提取液尽量少,使被测组分提取到小体积溶剂中,然后直接吸出适量供测定。 2、是挥去提取溶剂法 3、柱切换技术
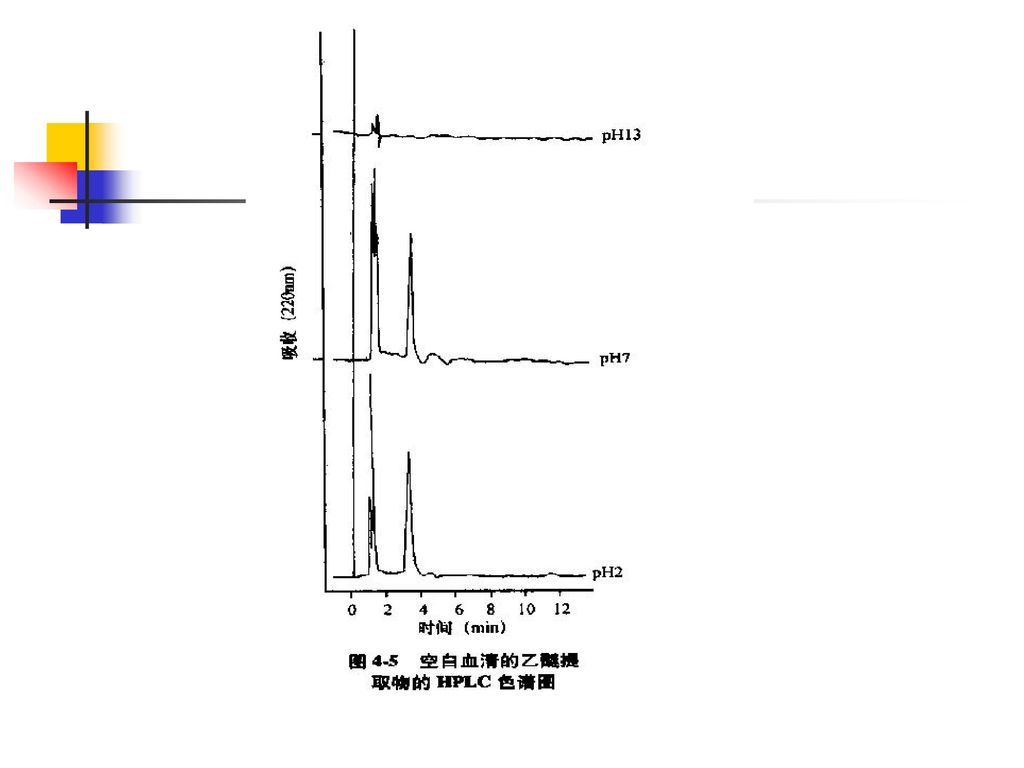
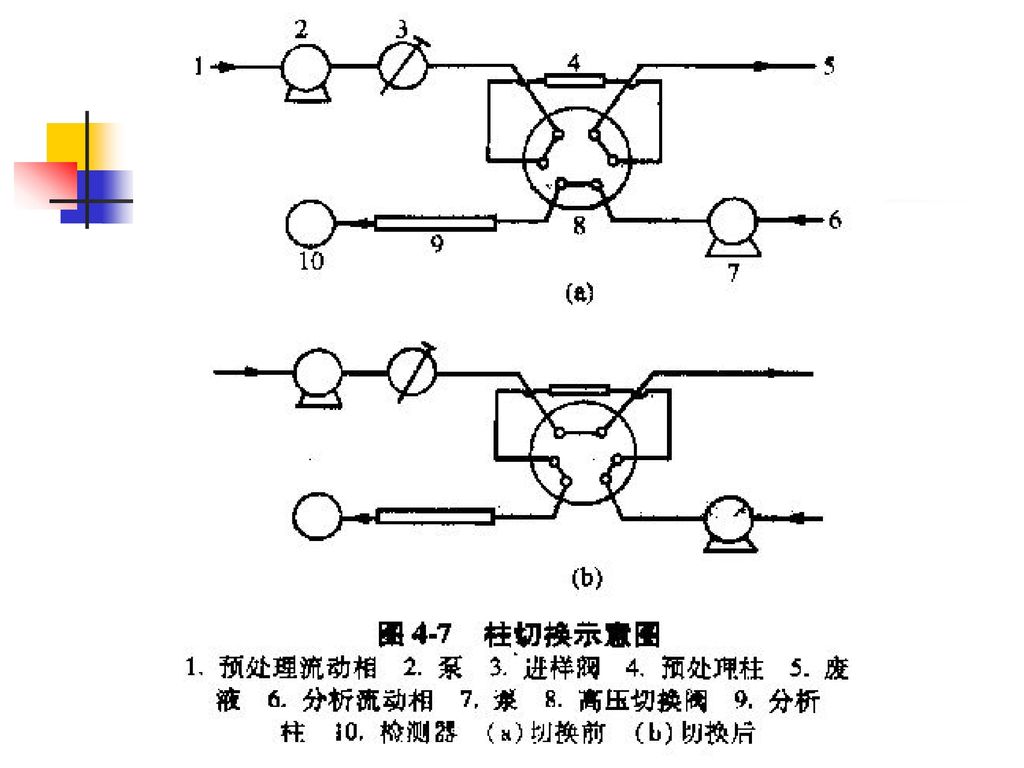
 分离、纯化与浓集. 提取法是应用最多的分离、纯化方法: 1、液-液提取法. 2、液-固提取法. 浓集的方法主要有: 1、是在末次提取时加入的提取液尽量少,使被测组分提取到小体积溶剂中,然后直接吸出适量供测定。 2、是挥去提取溶剂法. 3、柱切换技术.")
86
(四) 化学衍生化 目分离前药物进行化学衍生化的目的: 1、使药物变成具有能被分离的性质 2、 提高检测的灵敏度 3、增强药物的稳定行 4、提高对光学异构体分离的能力等 注: 药物分子中含有活泼H者均可被化学衍生化

87
三、定量分析方法验证 由于生物样品取样量少、药物浓度低、内源性物质的干扰及个体差异等多种因素影响生物样品测定,为了保证方法的可靠性,必须建立生物样品分析方法,并对方法进行验证。 (一) 特异性 (二) 标准曲线与线性范围 (三) 精密度与准确度 (四) 最低定量限 (五) 样品稳定性 (六) 提取回收率 (七) 质控样品 (八) 质量控制
 精密度与准确度. (四) 最低定量限. (五) 样品稳定性. (六) 提取回收率. (七) 质控样品. (八) 质量控制.")
88
2.根据实际生产水平: 硫酸长春新碱:原料用92.0~105.0% 注射用:90.0~110.0% 盐酸罂粟碱:原料用≥99.0% 片剂标示量:93.0~107.0% 注射剂标示量:95.0~105.0% 基因工程蛋白能:85~110% 3.根据主药含量多少: 主药含量最小:5μg,最大0.5g。 含量较大限度订为:95.0~105.0% 主药含量居中片剂订为:93.0~107.0% 主药含量较小订为:90 0~110.0或80.0~120 0%

89
系统适应性 1.理论塔板数:n= 2.分离度:R= 定量分析:R≥1.5 色谱柱尺寸;填料性能;流动相组成;柱外体积;积分参数
3.重复性(进样精密度) 外标法:对照品溶液(n ≥5) 内标法:相当于80%,100%,120%的对照品溶液,加入规定量内标溶液,分别至少进样3次(n ≥6) 峰面积RSD%≤2.0% 手动进样器:定量环(进样体积≥3×定量环体积) 自动进样器:
 外标法:对照品溶液(n ≥5) 内标法:相当于80%,100%,120%的对照品溶液,加入规定量内标溶液,分别至少进样3次(n ≥6) 峰面积RSD%≤2.0% 手动进样器:定量环(进样体积≥3×定量环体积) 自动进样器:")
90
系统适应性 4.拖尾因子: 色谱柱选择:封端柱 流动相:离子对色谱 进样量:色谱柱容量 积分参数 定量分析:Tf 0.95—1.05

91
Ch.P对HPLC法的要求 满足系统适应性的要求 塔板数:满足个论要求 分离度:Rs>1.5 进样精密度:2.0%
脱尾因子0.95<Tf<1.05 分离时间:10-30min 定量精密度:含量测定≤2.0%,痕量分析≤15%

92
常见的液相色谱参数的调整 流动相 —改变溶剂比例:色谱柱的含碳量 —添加剂:扫尾剂(有机胺,离子对试剂);缓冲液浓度(离子强度)
—pH:酸含量,缓冲对的缓冲容量 固定相 —封端柱 —pH耐受范围 —耐受高水相 柱温:升高柱温有利于增加传递速度,改善峰形和分离 进样量:在满足检测要求的前提下,尽量减少进样量 供试品溶剂:溶剂强度应低于流动相洗脱强度

Similar presentations











目的 证明所采用的分析方法适合于相应的检测要求,方法验证过程和结果均应记载在药品标准起草和修订说明中。>")









