Download presentation
Presentation is loading. Please wait.

1
第八章 (Ⅰ)杂环类药物的分 The Analysis of Heterocyclic drugs
杂环化合物:指环状有机化合物的碳环中夹杂有非碳元素原子(如O、S、N等)的化合物
的化合物.")
2
共性: (1)杂环类药物是合成药物中所占比例最多 的 一大类药物. (2)多为五元环或六元环,单环或并合环.
(3)杂环结构较稳定,不易开环,其性质受杂 原子种类、数目、位置影响. (4)杂环上取代基性质较活泼,常用于分析. (5)含氮杂环,其碱性的强弱往往用于分析.
杂环结构较稳定,不易开环,其性质受杂. 原子种类、数目、位置影响. (4)杂环上取代基性质较活泼,常用于分析. (5)含氮杂环,其碱性的强弱往往用于分析.")
3
第一节 吡啶类药物的分析 异烟肼(isoniazid) 吡啶(pyridine)
 吡啶(pyridine)")
4
尼可刹米(nikethamide)
")
5
异烟腙(ftivazide)
")
6
丙硫异烟胺(protionamide)
")
7
一、结构与性质 1. 吡啶环 弱碱性 pKb~8.8, 非水碱量法含量测定或沉淀反应鉴别 吡啶环可发生开环反应,可用于吡啶类药物的鉴别

8
2. 取代基: (1)异烟肼 位上酰肼基 弱酸性 非水酸量法 还原性 鉴别或氧化还原滴定法含量测定 可与某些羰基试剂发生缩合反应 鉴别或比色法含量测定 酰胺键易水解引入特殊杂质游离肼

9
(2)尼可刹米 位上酰胺基 易水解,遇碱水解后,释放出具有碱性的二乙胺,能使湿润的红色石蕊试纸变蓝色,故可以此进行鉴别。
尼可刹米 位上酰胺基 易水解,遇碱水解后,释放出具有碱性的二乙胺,能使湿润的红色石蕊试纸变蓝色,故可以此进行鉴别。")
10
(3)丙硫异烟胺 位上硫代甲酰胺基 易水解,加酸水解后,释放出硫化氢,能使湿润的醋酸铅试纸显黑色,故可以此进行鉴别。 3. UV和IR
丙硫异烟胺 位上硫代甲酰胺基 易水解,加酸水解后,释放出硫化氢,能使湿润的醋酸铅试纸显黑色,故可以此进行鉴别。 3. UV和IR")
11
二、鉴别试验 (一)吡啶环的反应 1. 沉淀反应 (1)与氯化汞的反应
吡啶环的反应 1. 沉淀反应 (1)与氯化汞的反应")
12
(2)与铜盐的反应 尼可刹米 ChP(2005) 【鉴别】(3)取本品 2 滴,加水 1 ml ,摇匀,加硫酸铜试液 2 滴与硫氰酸胺试液 3 滴,即生成草绿色沉淀。
与铜盐的反应 尼可刹米 ChP(2005) 【鉴别】(3)取本品 2 滴,加水 1 ml ,摇匀,加硫酸铜试液 2 滴与硫氰酸胺试液 3 滴,即生成草绿色沉淀。")
13
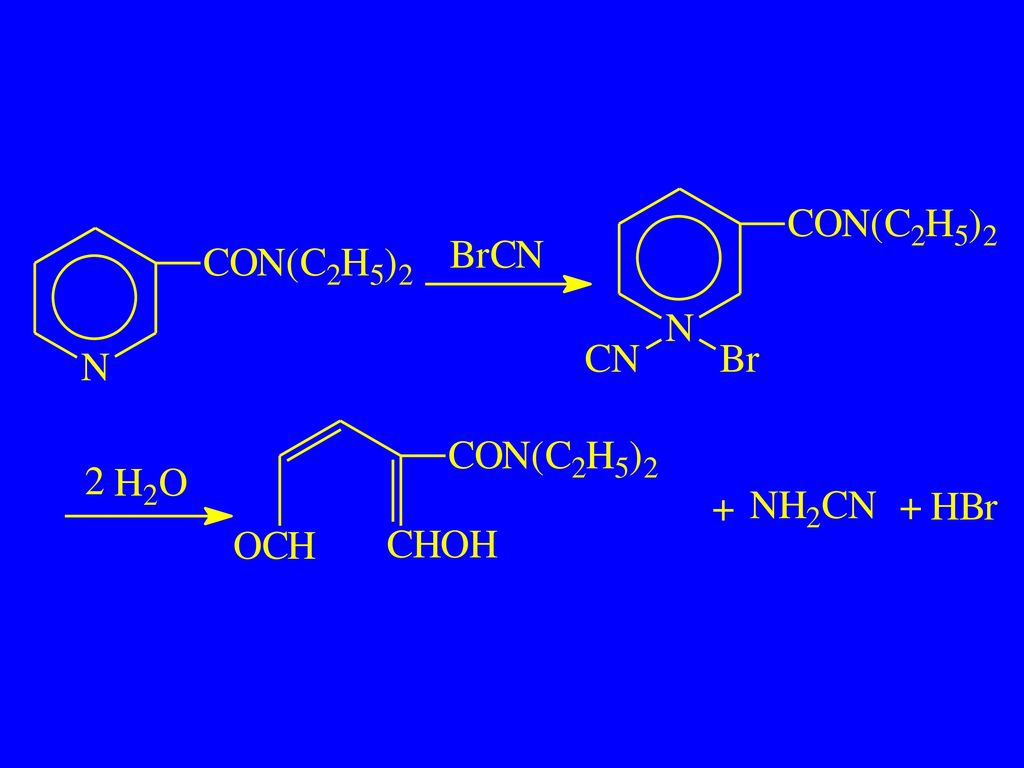
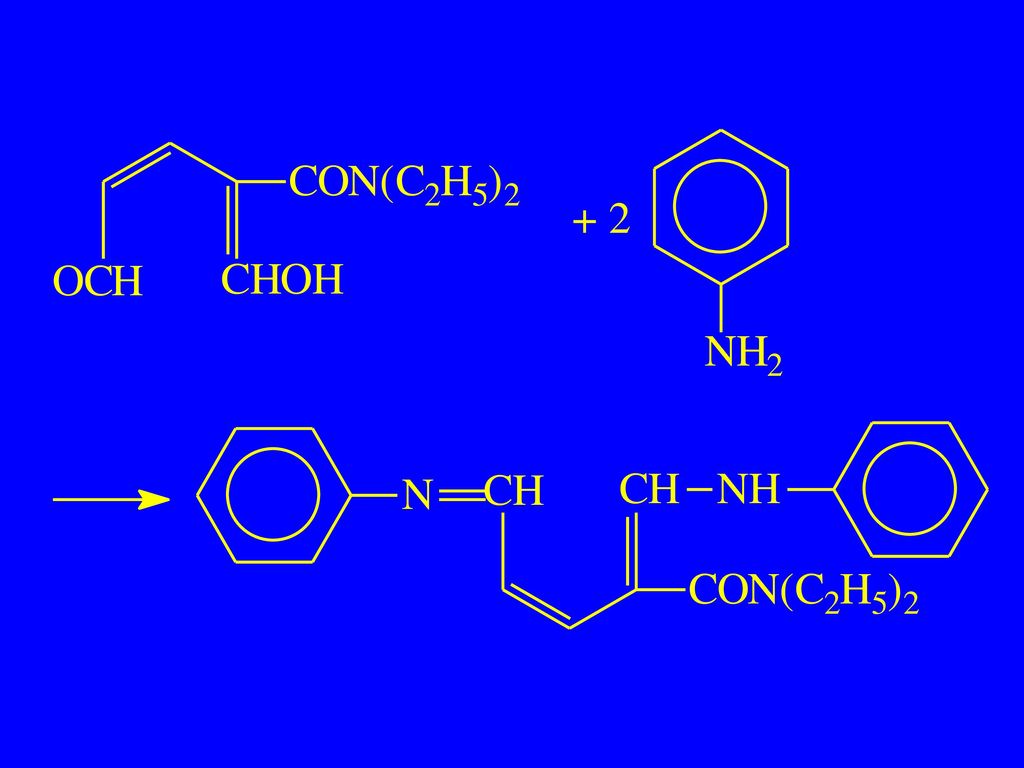
2. 吡啶环的开环反应 适用于吡啶 、′未取代,以及 、 为烷基或羧基的衍生物 (1)戊烯二醛反应(König反应)
戊烯二醛反应(König反应)")
14
尼可刹米 ChP(2005) 【鉴别】(2)取本品 1 滴,加水 50 ml,摇匀,分取 2 ml,加溴化氰试液 2 ml与 2.5% 苯胺溶液 3 ml,摇匀,溶液渐显黄色。
 【鉴别】(2)取本品 1 滴,加水 50 ml,摇匀,分取 2 ml,加溴化氰试液 2 ml与 2.5% 苯胺溶液 3 ml,摇匀,溶液渐显黄色。")
17
异烟肼:

18
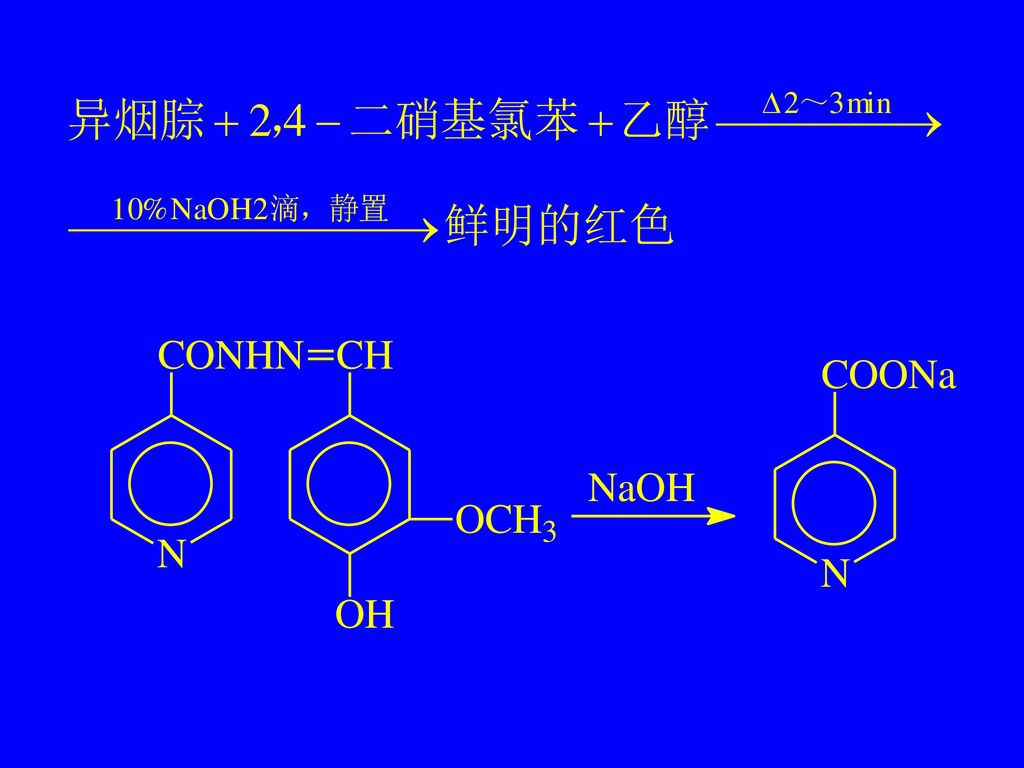
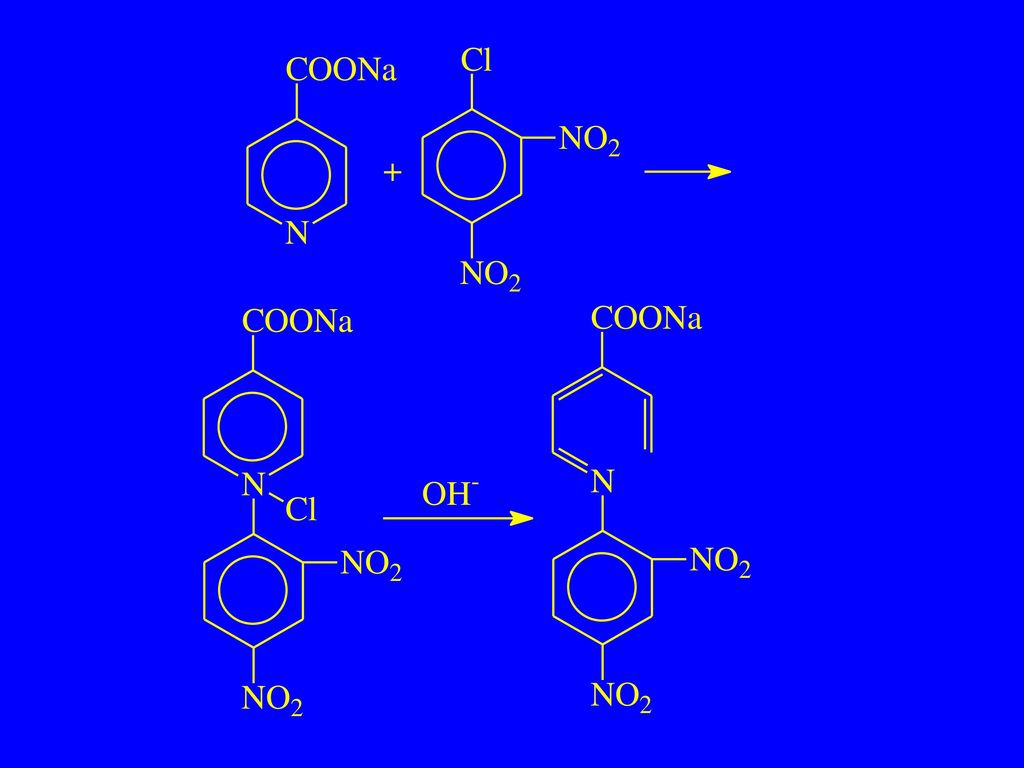
(2)二硝基氯苯反应 异烟腙 ChP(2005) 【鉴别】 取异烟腙约50 mg,加2,4-二硝基氯苯50 mg与乙醇3 m1,置水浴中煮沸2~3 min,加10%氢氧化钠溶液2滴,静置后,即显鲜明的红色。
二硝基氯苯反应 异烟腙 ChP(2005) 【鉴别】 取异烟腙约50 mg,加2,4-二硝基氯苯50 mg与乙醇3 m1,置水浴中煮沸2~3 min,加10%氢氧化钠溶液2滴,静置后,即显鲜明的红色。")
21
异烟肼不经处理的反应:

22
(二)酰肼基团的反应 1. 还原反应
酰肼基团的反应 1. 还原反应")
23
异烟肼 ChP(2005) 【鉴别】 (2)取异烟肼约10 mg,置试管中,加水2 ml溶解后,加氨制硝酸银试液1 m1,即发生气泡与黑色浑浊,并在试管壁上生成银镜。
 【鉴别】 (2)取异烟肼约10 mg,置试管中,加水2 ml溶解后,加氨制硝酸银试液1 m1,即发生气泡与黑色浑浊,并在试管壁上生成银镜。")
24
2. 缩合反应

25
异烟肼 ChP(2005) 【鉴别】 (1)取本品约0.1 g,加水5 ml溶解后,加10%香草醛的乙醇溶液1 m1,摇匀,微热,放冷,即析出黄色结晶,滤过,用稀乙醇重结晶,在105℃干燥后,测定熔点,其熔点为228~231℃,熔融时同时分解。
 【鉴别】 (1)取本品约0.1 g,加水5 ml溶解后,加10%香草醛的乙醇溶液1 m1,摇匀,微热,放冷,即析出黄色结晶,滤过,用稀乙醇重结晶,在105℃干燥后,测定熔点,其熔点为228~231℃,熔融时同时分解。")
26
(三)分解产物的反应 1. 尼可刹米 ChP(2005) 【鉴别】 (1)取本品 10 滴,加氢氧化钠试液 3 ml ,加热,即发生二乙胺的臭气,能使湿润的红色石蕊试纸变蓝色。
分解产物的反应 1. 尼可刹米 ChP(2005) 【鉴别】 (1)取本品 10 滴,加氢氧化钠试液 3 ml ,加热,即发生二乙胺的臭气,能使湿润的红色石蕊试纸变蓝色。")
27
2. 丙硫异烟胺 ChP(2005) 【鉴别】(1)取本品约 50 mg,加盐酸溶液(9→100)3 ml ,缓缓加热,发生的气体可使湿润的醋酸铅试纸显黑色。
 【鉴别】(1)取本品约 50 mg,加盐酸溶液(9→100)3 ml ,缓缓加热,发生的气体可使湿润的醋酸铅试纸显黑色。")
28
(四)紫外吸收光谱特征 丙硫异烟胺 ChP(2005) 【鉴别】(1)取本品,加乙醇制成每1 ml 中含 20 g 的溶液,照分光光度法(附录ⅣA)测定,在 291 nm 的波长处有最大吸收,吸收度约为0.78。
紫外吸收光谱特征 丙硫异烟胺 ChP(2005) 【鉴别】(1)取本品,加乙醇制成每1 ml 中含 20 g 的溶液,照分光光度法(附录ⅣA)测定,在 291 nm 的波长处有最大吸收,吸收度约为0.78。")
29
(五)IR 尼可刹米 ChP(2005) 【鉴别】(4)本品的红外吸收图谱应与对照的图谱(光谱集 135 图)一致。
IR 尼可刹米 ChP(2005) 【鉴别】(4)本品的红外吸收图谱应与对照的图谱(光谱集 135 图)一致。")
30
三、杂质检查 (一)异烟肼中游离肼的检查 杂质来源 原料引入、降解产生 1. 薄层色谱法(TLC) (1) ChP(2005) 杂质对照品法
异烟肼中游离肼的检查 杂质来源 原料引入、降解产生 1. 薄层色谱法(TLC) (1) ChP(2005) 杂质对照品法")
31
取本品,加水制成每 1ml 中含 50mg的溶液,作为供试品溶液。另取硫酸肼加水制成每 1ml中含 0
取本品,加水制成每 1ml 中含 50mg的溶液,作为供试品溶液。另取硫酸肼加水制成每 1ml中含 0.20mg(相当于游离肼50µg 的溶液,作为对照溶液。吸取供试品溶液 10µl 与对照溶液 2µl 分别点于同一硅胶薄层板(用羧甲基纤维素钠溶液制备)上,以异丙醇-丙酮(3∶2)为展开剂,展开后,晾干,喷以乙醇制对二甲氨基苯甲醛试液,15min后检视,在供试品主斑点前方与硫酸肼斑点相应的位置上,不得显黄色斑点。
上,以异丙醇-丙酮(3∶2)为展开剂,展开后,晾干,喷以乙醇制对二甲氨基苯甲醛试液,15min后检视,在供试品主斑点前方与硫酸肼斑点相应的位置上,不得显黄色斑点。")
32
TLC法(ChP) 杂质对照品法 ①样品溶液 异烟肼的水溶液(50mg/ml) ②对照溶液 硫酸肼加水制成每ml 0.20 mg
(相当于游离肼50ug)溶液 ③分离系统(展开剂,流动相) 异丙醇-丙酮(3∶2) ④点样量 样品液:10l 对照液:2l
溶液. ③分离系统(展开剂,流动相) 异丙醇-丙酮(3∶2) ④点样量. 样品液:10l 对照液:2l.")
33
× O 鲜黄色 棕橙色

34
(2) BP(1998) 游离肼 杂质对照品法 有关物质 高低浓度对比法
 BP(1998) 游离肼 杂质对照品法 有关物质 高低浓度对比法")
35
TLC法(BP) 杂质对照品法+自身对照法
(1)样品溶液 异烟肼的丙酮-水(1 ∶1 )溶液(100mg/ml) (2)对照溶液 硫酸肼的丙酮-水(1 ∶1 )溶液(0.05 mg/ml ) 异烟肼的丙酮-水(1 ∶1 )溶液(0.2mg/ml) (3)分离系统 乙酸乙酯-丙酮-甲醇-水(50∶20 ∶20 ∶ 10) (4)点样量 样品液:5l 对照液:5l
样品溶液. 异烟肼的丙酮-水(1 ∶1 )溶液(100mg/ml) (2)对照溶液. 硫酸肼的丙酮-水(1 ∶1 )溶液(0.05 mg/ml ) 异烟肼的丙酮-水(1 ∶1 )溶液(0.2mg/ml) (3)分离系统. 乙酸乙酯-丙酮-甲醇-水(50∶20 ∶20 ∶ 10) (4)点样量. 样品液:5l 对照液:5l.")
36
2. 比浊法 JP(13) 检查方法 灵敏度法 反应原理 游离肼+水杨醛→水杨醛腙↓ 优点:不用对照品,价廉、简单易行 缺点:准确度差
 检查方法 灵敏度法 反应原理 游离肼+水杨醛→水杨醛腙↓ 优点:不用对照品,价廉、简单易行 缺点:准确度差")
37
3. 差示分光光度法 游离肼+对二甲氨基苯甲醛→黄色缩合物 于456nm波长处有最大吸收
游离肼+3%丙酮→无色的二甲基甲酮连氮

38
参比:异烟肼+游离肼+对二甲氨基苯甲醛+3%丙酮
样品:异烟肼+游离肼+对二甲氨基苯甲醛 测定,对照法定量

39
(二)尼可刹米中有关物质的检查 ChP(2005) TLC 高低浓度对比法 配制两种不同浓度的对照溶液
尼可刹米中有关物质的检查 ChP(2005) TLC 高低浓度对比法 配制两种不同浓度的对照溶液")
40
供试品 对照品 供试液 对照液

41
(三)硝苯地平中有关物质的检查 HPLC 供试品自身对照法、 有关杂质对照品法 (ChP USP) 有关杂质对照品A+B
硝苯地平中有关物质的检查 HPLC 供试品自身对照法、 有关杂质对照品法 (ChP USP) 有关杂质对照品A+B")
42
Nifedipine C17H18N2O6

43
Specified impurities A, B, C, D. BP2007
A. R = NO2: dimethyl 2,6-dimethyl-4-(2-nitrophenyl)pyridine-3,5-dicarboxylate (nitrophenylpyridine analogue), B. R = NO: dimethyl 2,6-dimethyl-4-(2-nitrosophenyl)pyridine-3,5-dicarboxylate (nitrosophenylpyridine analogue),
pyridine-3,5-dicarboxylate (nitrophenylpyridine analogue), B. R = NO: dimethyl 2,6-dimethyl-4-(2-nitrosophenyl)pyridine-3,5-dicarboxylate (nitrosophenylpyridine analogue),")
44
C D

45
四、含量测定 (一)异烟肼的含量测定 1. 氧化还原滴定法 (1)溴酸钾法 ChP(2005)原料及制剂
异烟肼的含量测定 1. 氧化还原滴定法 (1)溴酸钾法 ChP(2005)原料及制剂")
46
反应摩尔比 3∶2

47
取本品约 0.2g,精密称定,置 100ml量瓶中,加水使溶解并稀释至刻度,摇匀;精密量取25m1,加水50m1、盐酸20ml与甲基橙指示剂1滴,用溴酸钾滴定液( mo1/L)缓缓滴定(温度保持在18~25℃)至粉红色消失。每1ml的溴酸钾滴定液( mol/L)相当于3.429mg的C6H7N3O。
缓缓滴定(温度保持在18~25℃)至粉红色消失。每1ml的溴酸钾滴定液( mol/L)相当于3.429mg的C6H7N3O。")
48
(2)溴量法 反应摩尔比 1∶4
溴量法 反应摩尔比 1∶4")
49
(3)剩余碘量法 反应摩尔比 1∶4
剩余碘量法 反应摩尔比 1∶4")
50
2. 非水溶液滴定法 (1)非水酸量法 (2)非水碱量法
非水酸量法 (2)非水碱量法")
51
(二)尼可刹米的含量测定 1. 非水溶液滴定法 ChP(2005)原料 2. 紫外分光光度法 ChP(2005)注射液 内容量移液管
尼可刹米的含量测定 1. 非水溶液滴定法 ChP(2005)原料 2. 紫外分光光度法 ChP(2005)注射液 内容量移液管")
52
用内容量移液管精密量取本品1m1,置 100ml 量瓶中,用 0.5% 硫酸溶液分次洗涤移液管内壁,洗液并入量瓶中,加
0.5%硫酸溶液稀释至刻度,摇匀,精密量取适量,加0.5%硫酸溶液稀释成每1ml中约含尼可刹米20µg的溶液,在263nm的波长处测定吸收度,按C10H14N2O的吸收系数( )为292计算,即得。
为292计算,即得。")
53
(三)异烟腙的含量测定 非水溶液滴定法 电位法指示终点 ChP(2005)原料及片剂
异烟腙的含量测定 非水溶液滴定法 电位法指示终点 ChP(2005)原料及片剂")
54
(四)丙硫异烟胺的含量测定 1. 非水溶液滴定法 ChP(2005)原料 2. 银量法 ChP(2005)片剂
丙硫异烟胺的含量测定 1. 非水溶液滴定法 ChP(2005)原料 2. 银量法 ChP(2005)片剂")
55
取本品20片,精密称定,研细,精密称取适量(约相当于丙硫异烟胺0
取本品20片,精密称定,研细,精密称取适量(约相当于丙硫异烟胺0.3g)置具塞锥形瓶中,加丙酮20ml使丙硫异烟胺溶解,精密加入硝酸银滴定液(0.1mo1/L)50m1,摇匀,放置15min,加水50m1、硝酸3m1、硝基苯5ml与硫酸铁铵指示液2m1,用硫氰酸铵滴定液(0.1mo1/L)滴定,并将滴定的结果用空白试验校正。每lml的硝酸银滴定液(0.1mo1/L)相当于 9.014mg的C9H12N2S。
置具塞锥形瓶中,加丙酮20ml使丙硫异烟胺溶解,精密加入硝酸银滴定液(0.1mo1/L)50m1,摇匀,放置15min,加水50m1、硝酸3m1、硝基苯5ml与硫酸铁铵指示液2m1,用硫氰酸铵滴定液(0.1mo1/L)滴定,并将滴定的结果用空白试验校正。每lml的硝酸银滴定液(0.1mo1/L)相当于 mg的C9H12N2S。")
56
第四节 吩噻嗪类药物的分析 吩噻嗪类药物的基本结构

57
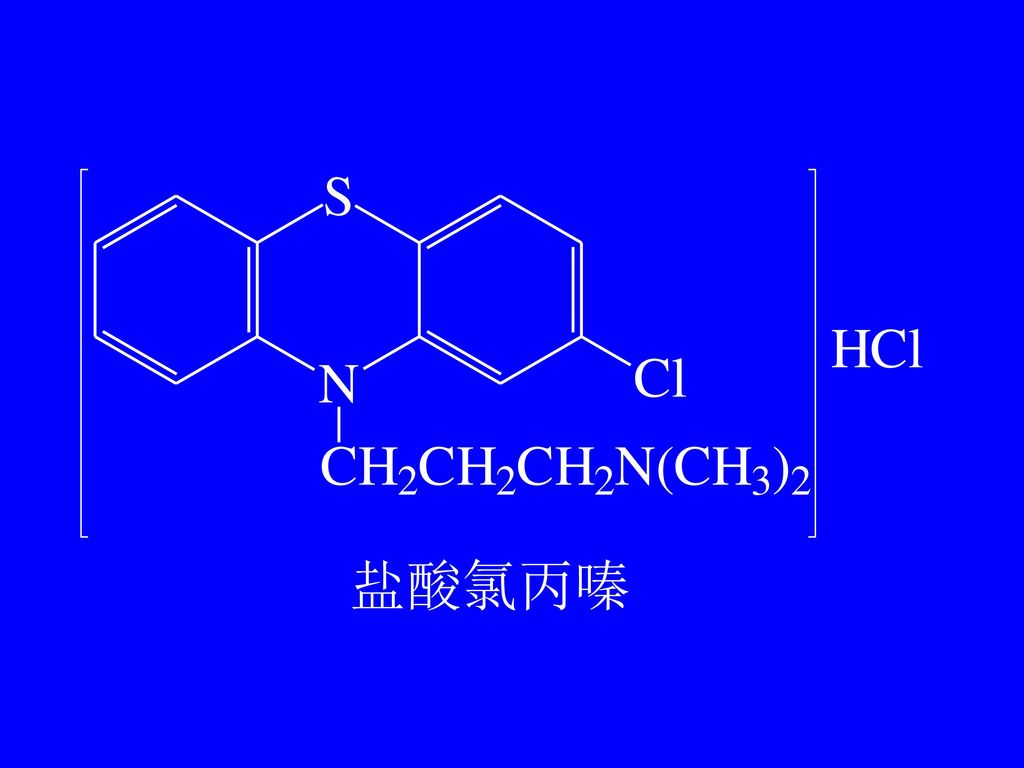
不同点:R,R’取代基不同 R R’ 盐类 药名 盐酸丙嗪 -(CH2)3N(CH3)2 -H HCl -Cl 盐酸氯丙嗪 盐酸异丙嗪
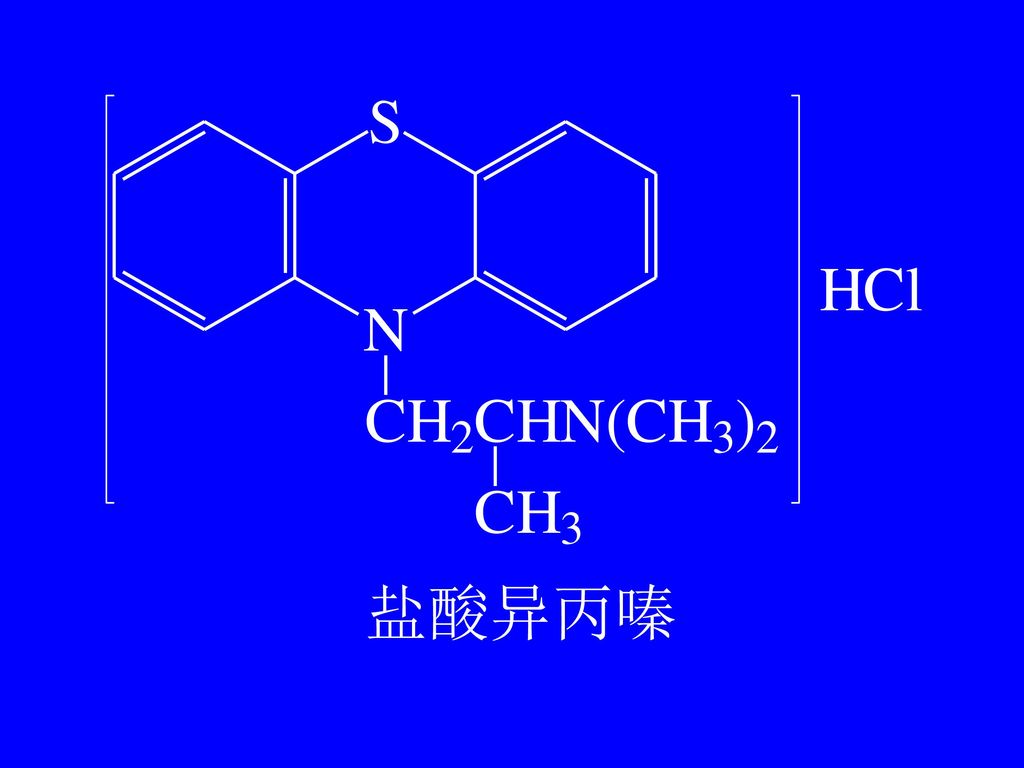
-CH2CH(CH3)N(CH3)2 盐酸异丙嗪
N(CH3)2. 盐酸异丙嗪.")
60
奋乃静

61
癸氟奋乃静

62
盐酸氟奋乃静

63
盐酸三氟拉嗪

64
盐酸二氧丙嗪

65
一、结构与性质 (一)结构分析 1. 硫氮杂蒽母核 (1)含S、N的三环共轭的大体系,S、N与苯环形成p-共轭——具有紫外吸收光谱特征
结构分析 1. 硫氮杂蒽母核 (1)含S、N的三环共轭的大体系,S、N与苯环形成p-共轭——具有紫外吸收光谱特征")
66
(2)硫氮杂蒽环上硫原子为-2价,具有还原性,易氧化呈色
(3)硫氮杂蒽环上硫原子有两对孤对电子,易与金属离子络合呈色
硫氮杂蒽环上硫原子有两对孤对电子,易与金属离子络合呈色.")
67
2. 取代基 R:脂烃胺基、哌嗪基,具碱性 R:卤素

68
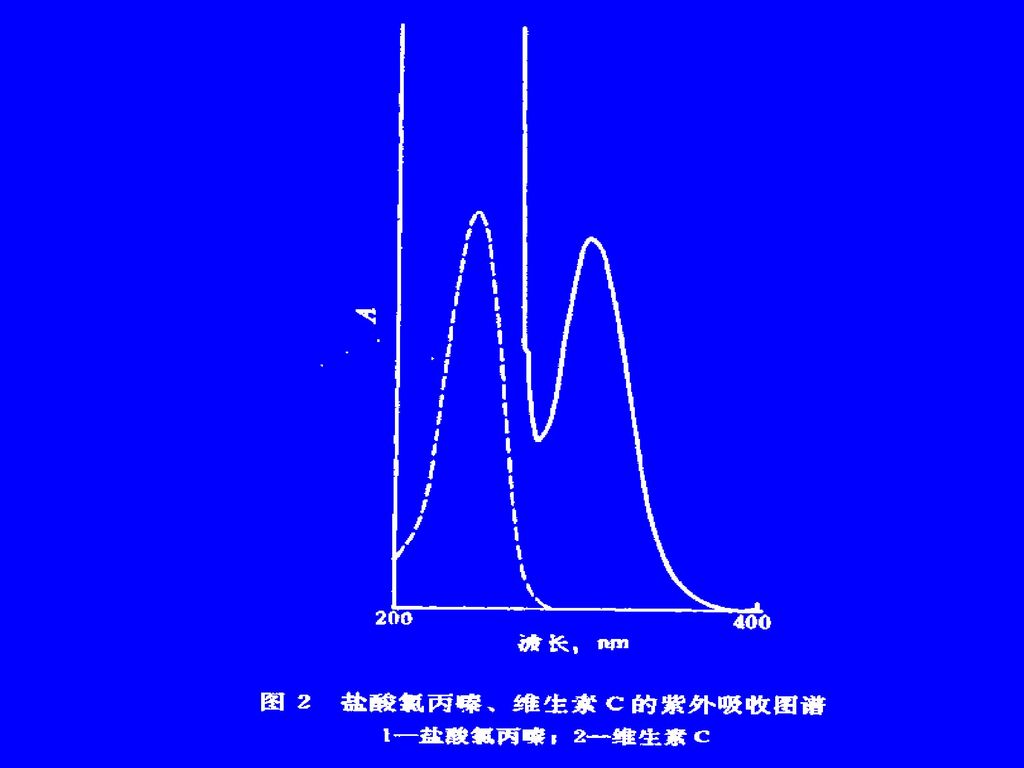
(二)性质 1. 紫外与红外吸收光谱 本类药物的紫外特征吸收,主要由母核三环的π系统所产生,一般具有三个峰值。即在 204~209nm(205nm附近)、250~265nm(254nm附近)和300~325nm(300nm附近)。最强峰多在 250~265nm。
、250~265nm(254nm附近)和300~325nm(300nm附近)。最强峰多在 250~265nm。 .")
69
204~209nm、250~265nm(强)、300~325nm
、300~325nm")
70
2位上的取代基(R′)不同,会引起吸收峰发生位移。
其结构中-2 价的硫,易氧化,氧化产物砜及亚砜有四个吸收峰。 吩噻嗪类药物随取代基 R 和R′的不同,产生不同的红外吸收光谱。

71
2. 易被氧化呈色 3. 易与金属离子络合呈色 4. 碱性 未被氧化的S + Pd 2+ 有色配位化合物 砜(亚砜)+ Pd 2+
+ Pd 2+")
72
二、鉴别试验 (一)UV和IR 奋乃静 ChP(2005)
[鉴别] (2)取本品,加无水乙醇制成每1ml 中含 7g 的溶液,照分光光度法(附录ⅣA)测定,在 258nm 的波长处有最大吸收,吸收度约为0.65。
取本品,加无水乙醇制成每1ml 中含 7g 的溶液,照分光光度法(附录ⅣA)测定,在 258nm 的波长处有最大吸收,吸收度约为0.65。")
73
(3)本品的红外吸收图谱应与对照的图谱(光谱集 243 图)一致
本品的红外吸收图谱应与对照的图谱(光谱集 243 图)一致")
74
(二) 显色反应 1. 氧化剂氧化显色 氧化剂 硫酸、硝酸、过氧化氢 奋乃静 ChP(2005) [鉴别] (1)取本品 5mg,加盐酸与水各 1ml,加热至80℃,加入过氧化氢溶液数滴,即显深红色;放置后,红色渐褪去。
 [鉴别] (1)取本品 5mg,加盐酸与水各 1ml,加热至80℃,加入过氧化氢溶液数滴,即显深红色;放置后,红色渐褪去。 .")
75
盐酸异丙嗪 ChP(2005) [鉴别] (1)取本品约 5mg,加硫酸5ml 溶解后,溶液显樱桃红色;放置后,色渐变深。 (2)取本品约 0.1g,加水 3ml 溶解后,加硝酸 1ml,即生成红色沉淀;加热,沉淀即溶解,溶液由红色转变为橙黄色。
![盐酸异丙嗪 ChP(2005) [鉴别] (1)取本品约 5mg,加硫酸5ml 溶解后,溶液显樱桃红色;放置后,色渐变深。 (2)取本品约 0.1g,加水 3ml 溶解后,加硝酸 1ml,即生成红色沉淀;加热,沉淀即溶解,溶液由红色转变为橙黄色。](http://slidesplayer.com/slide/11456523/61/images/75/%E7%9B%90%E9%85%B8%E5%BC%82%E4%B8%99%E5%97%AA+ChP%EF%BC%882005%EF%BC%89+%5B%E9%89%B4%E5%88%AB%5D+%EF%BC%881%EF%BC%89%E5%8F%96%E6%9C%AC%E5%93%81%E7%BA%A6+5mg%EF%BC%8C%E5%8A%A0%E7%A1%AB%E9%85%B85ml+%E6%BA%B6%E8%A7%A3%E5%90%8E%EF%BC%8C%E6%BA%B6%E6%B6%B2%E6%98%BE%E6%A8%B1%E6%A1%83%E7%BA%A2%E8%89%B2%EF%BC%9B%E6%94%BE%E7%BD%AE%E5%90%8E%EF%BC%8C%E8%89%B2%E6%B8%90%E5%8F%98%E6%B7%B1%E3%80%82+%EF%BC%882%EF%BC%89%E5%8F%96%E6%9C%AC%E5%93%81%E7%BA%A6+0.1g%EF%BC%8C%E5%8A%A0%E6%B0%B4+3ml+%E6%BA%B6%E8%A7%A3%E5%90%8E%EF%BC%8C%E5%8A%A0%E7%A1%9D%E9%85%B8+1ml%EF%BC%8C%E5%8D%B3%E7%94%9F%E6%88%90%E7%BA%A2%E8%89%B2%E6%B2%89%E6%B7%80%EF%BC%9B%E5%8A%A0%E7%83%AD%EF%BC%8C%E6%B2%89%E6%B7%80%E5%8D%B3%E6%BA%B6%E8%A7%A3%EF%BC%8C%E6%BA%B6%E6%B6%B2%E7%94%B1%E7%BA%A2%E8%89%B2%E8%BD%AC%E5%8F%98%E4%B8%BA%E6%A9%99%E9%BB%84%E8%89%B2%E3%80%82.jpg "盐酸异丙嗪 ChP(2005) [鉴别] (1)取本品约 5mg,加硫酸5ml 溶解后,溶液显樱桃红色;放置后,色渐变深。 (2)取本品约 0.1g,加水 3ml 溶解后,加硝酸 1ml,即生成红色沉淀;加热,沉淀即溶解,溶液由红色转变为橙黄色。")
76
2. 与钯离子络合显色 癸氟奋乃静 ChP(2005) [鉴别] (2)取本品约 5mg,加甲醇2ml 溶解后,加 0.1% 氯化钯溶液 3ml,即有沉淀生成,并显红色,再加过量的氯化钯溶液,颜色变深。
 [鉴别] (2)取本品约 5mg,加甲醇2ml 溶解后,加 0.1% 氯化钯溶液 3ml,即有沉淀生成,并显红色,再加过量的氯化钯溶液,颜色变深。 .")
77
(三)分解产物的反应 癸氟奋乃静 ChP(2005) [鉴别] (1)取本品 15 ~20mg,加碳酸钠与碳酸钾各约0.1g,混匀,在 600℃ 炽灼 15~20分钟,放冷,加水2ml 使溶解,加盐酸溶液(1→2)酸化,滤过,滤液加茜素锆试液 0.5ml,应显黄色。
![(三)分解产物的反应 癸氟奋乃静 ChP(2005) [鉴别] (1)取本品 15 ~20mg,加碳酸钠与碳酸钾各约0.1g,混匀,在 600℃ 炽灼 15~20分钟,放冷,加水2ml 使溶解,加盐酸溶液(1→2)酸化,滤过,滤液加茜素锆试液 0.5ml,应显黄色。](http://slidesplayer.com/slide/11456523/61/images/77/%EF%BC%88%E4%B8%89%EF%BC%89%E5%88%86%E8%A7%A3%E4%BA%A7%E7%89%A9%E7%9A%84%E5%8F%8D%E5%BA%94+%E7%99%B8%E6%B0%9F%E5%A5%8B%E4%B9%83%E9%9D%99+ChP%EF%BC%882005%EF%BC%89+%5B%E9%89%B4%E5%88%AB%5D+%EF%BC%881%EF%BC%89%E5%8F%96%E6%9C%AC%E5%93%81+15+%EF%BD%9E20mg%EF%BC%8C%E5%8A%A0%E7%A2%B3%E9%85%B8%E9%92%A0%E4%B8%8E%E7%A2%B3%E9%85%B8%E9%92%BE%E5%90%84%E7%BA%A60.1g%EF%BC%8C%E6%B7%B7%E5%8C%80%EF%BC%8C%E5%9C%A8+600%E2%84%83+%E7%82%BD%E7%81%BC+15%EF%BD%9E20%E5%88%86%E9%92%9F%EF%BC%8C%E6%94%BE%E5%86%B7%EF%BC%8C%E5%8A%A0%E6%B0%B42ml+%E4%BD%BF%E6%BA%B6%E8%A7%A3%EF%BC%8C%E5%8A%A0%E7%9B%90%E9%85%B8%E6%BA%B6%E6%B6%B2%EF%BC%881%E2%86%922%EF%BC%89%E9%85%B8%E5%8C%96%EF%BC%8C%E6%BB%A4%E8%BF%87%EF%BC%8C%E6%BB%A4%E6%B6%B2%E5%8A%A0%E8%8C%9C%E7%B4%A0%E9%94%86%E8%AF%95%E6%B6%B2+0.5ml%EF%BC%8C%E5%BA%94%E6%98%BE%E9%BB%84%E8%89%B2%E3%80%82.jpg "(三)分解产物的反应 癸氟奋乃静 ChP(2005) [鉴别] (1)取本品 15 ~20mg,加碳酸钠与碳酸钾各约0.1g,混匀,在 600℃ 炽灼 15~20分钟,放冷,加水2ml 使溶解,加盐酸溶液(1→2)酸化,滤过,滤液加茜素锆试液 0.5ml,应显黄色。")
79
(四)氯化物的反应 盐酸氯丙嗪 ChP(2005) [鉴别] (3)本品的水溶液显氯化物的鉴别反应(附录 Ⅲ)。
![(四)氯化物的反应 盐酸氯丙嗪 ChP(2005) [鉴别] (3)本品的水溶液显氯化物的鉴别反应(附录 Ⅲ)。](http://slidesplayer.com/slide/11456523/61/images/79/%EF%BC%88%E5%9B%9B%EF%BC%89%E6%B0%AF%E5%8C%96%E7%89%A9%E7%9A%84%E5%8F%8D%E5%BA%94+%E7%9B%90%E9%85%B8%E6%B0%AF%E4%B8%99%E5%97%AA+ChP%EF%BC%882005%EF%BC%89+%5B%E9%89%B4%E5%88%AB%5D+%EF%BC%883%EF%BC%89%E6%9C%AC%E5%93%81%E7%9A%84%E6%B0%B4%E6%BA%B6%E6%B6%B2%E6%98%BE%E6%B0%AF%E5%8C%96%E7%89%A9%E7%9A%84%E9%89%B4%E5%88%AB%E5%8F%8D%E5%BA%94%EF%BC%88%E9%99%84%E5%BD%95+%E2%85%A2%EF%BC%89%E3%80%82.jpg "(四)氯化物的反应 盐酸氯丙嗪 ChP(2005) [鉴别] (3)本品的水溶液显氯化物的鉴别反应(附录 Ⅲ)。")
80
三、有关物质检查 1. 癸氟奋乃静及其注射液 TLC 参比杂质对照品法 以盐酸氟奋乃静为对照品 2. 其他药物 TLC 高低浓度对比法

81
癸氟奋乃静有关物质 取本品,加甲醇溶解,制成每1ml 中含25mg的溶液,作为供 试品溶液。另取盐酸氟奋乃静对照品适量,加甲醇稀释制成每1ml 中含0.50mg的溶液, 作为对照品溶液。照薄层色谱法(附录Ⅴ B)试验,取上述两种溶液各20μl ,分别点 于同一硅胶GF254 薄层板上,以丙酮-环己烷-浓氨溶液(80:30:5) 为展开剂,展开 后,晾干,先置紫外光灯(254nm) 下检视;再喷以硫酸溶液(1→2)后检视。供试品溶液 如显杂质斑点,与对照品溶液的主斑点比较,均不得更深。
试验,取上述两种溶液各20μl ,分别点 于同一硅胶GF254 薄层板上,以丙酮-环己烷-浓氨溶液(80:30:5) 为展开剂,展开 后,晾干,先置紫外光灯(254nm) 下检视;再喷以硫酸溶液(1→2)后检视。供试品溶液 如显杂质斑点,与对照品溶液的主斑点比较,均不得更深。")
82
四、含量测定 (一) 非水溶液滴定法 原料药 存在问题:
(一) 非水溶液滴定法 原料药 存在问题: HClO4在冰醋酸溶液中,具氧化性,可氧化吩噻嗪类药物产生红色的氧化物,干扰指示剂终点的观察。排除方法: (1)改用中性溶剂 (2)加抗坏血酸 (3)电位法指示终点
 非水溶液滴定法 原料药. 存在问题: HClO4在冰醋酸溶液中,具氧化性,可氧化吩噻嗪类药物产生红色的氧化物,干扰指示剂终点的观察。排除方法: (1)改用中性溶剂. (2)加抗坏血酸. (3)电位法指示终点.")
83
滴定液 溶剂 指示剂 盐酸氯丙嗪 高氯酸 醋酐 橙黄Ⅳ 奋乃静 高氯酸 冰醋酸 结晶紫 奋乃静注射液 碱化游离→氯仿提取 →挥干氯仿→高氯酸滴定

84
(二)紫外分光光度法 1. 直接分光光度法 盐酸异丙嗪:片剂测定波长为249nm( 为910);注射液为了消除抗氧剂维生素C(max 243nm)对测定的干扰,测定波长改为299nm( 为108)。
;注射液为了消除抗氧剂维生素C(max 243nm)对测定的干扰,测定波长改为299nm( 为108)。 .")
85
同理,盐酸氯丙嗪片剂测定波长为254nm( 为915);注射液测定波长改为306nm( 为115)。
;注射液测定波长改为306nm( 为115)。")
87
2. 萃取后分光光度法 盐酸氯丙嗪注射液

88
3. 二阶导数分光光度法 盐酸氯丙嗪注射液 抗氧剂维生素C的二阶导数光谱近似为接近基线的一条直线,不干扰盐酸氯丙嗪的测定

89
因此盐酸氯丙嗪可从其二阶导数光谱量取峰266nm~谷254nm距离,标准曲线法定量

90
4. 萃取—双波长分光光度法 (1)原理 在待测组分(a)的最大吸收波长(测定波长,1)处测定待测组分和干扰组分(b)吸收度的总和;另选一适当波长(参比波长,2)测定吸收度,并使干扰组分在测定波长和参比波长处的吸收度相等,即 ,而待测组分在这两个波长处吸收度的差值足够大。
的最大吸收波长(测定波长,1)处测定待测组分和干扰组分(b)吸收度的总和;另选一适当波长(参比波长,2)测定吸收度,并使干扰组分在测定波长和参比波长处的吸收度相等,即 ,而待测组分在这两个波长处吸收度的差值足够大。")
91
(2)定量依据 样品在二波长下吸收度差值(A):
定量依据 样品在二波长下吸收度差值(A):")
92
即,吸收度差值(A)仅与待测组分的浓度有关,而与干扰组分无关,干扰组分的干扰被消除。
仅与待测组分的浓度有关,而与干扰组分无关,干扰组分的干扰被消除。")
93
(3)必要条件 ① 干扰组分在两个波长处吸收度相等 ② 待测组分在两个波长处吸收度相差足够大
必要条件 ① 干扰组分在两个波长处吸收度相等 ② 待测组分在两个波长处吸收度相差足够大")
94
(4)应用 ①萃取 除去抗氧剂的干扰
应用 ①萃取 除去抗氧剂的干扰")
95
②双波长分光光度法测定 排除氧化产物的干扰 测定波长 nm 参比波长 nm 对照法定量

96
(三) 铈量法 (吩噻嗪类、硝苯地平) 自身指示终点或电位法、永停法指示终点
 铈量法 (吩噻嗪类、硝苯地平) 自身指示终点或电位法、永停法指示终点")
97
优点: 硫酸铈作滴定剂具有高的氧化电位,且为一价还原;不存在对环取代基的副反应,专属性高。 本法既可用于原料,亦可用于片剂的测定

98
(四)比色法 1. 钯离子比色法 反应在 pH2±0.1 的缓冲液中进行
比色法 1. 钯离子比色法 反应在 pH2±0.1 的缓冲液中进行")
99
优点: 钯离子比色法可选择性地用于未被氧化的吩噻嗪类药物的测定 2. 铁盐比色法

100
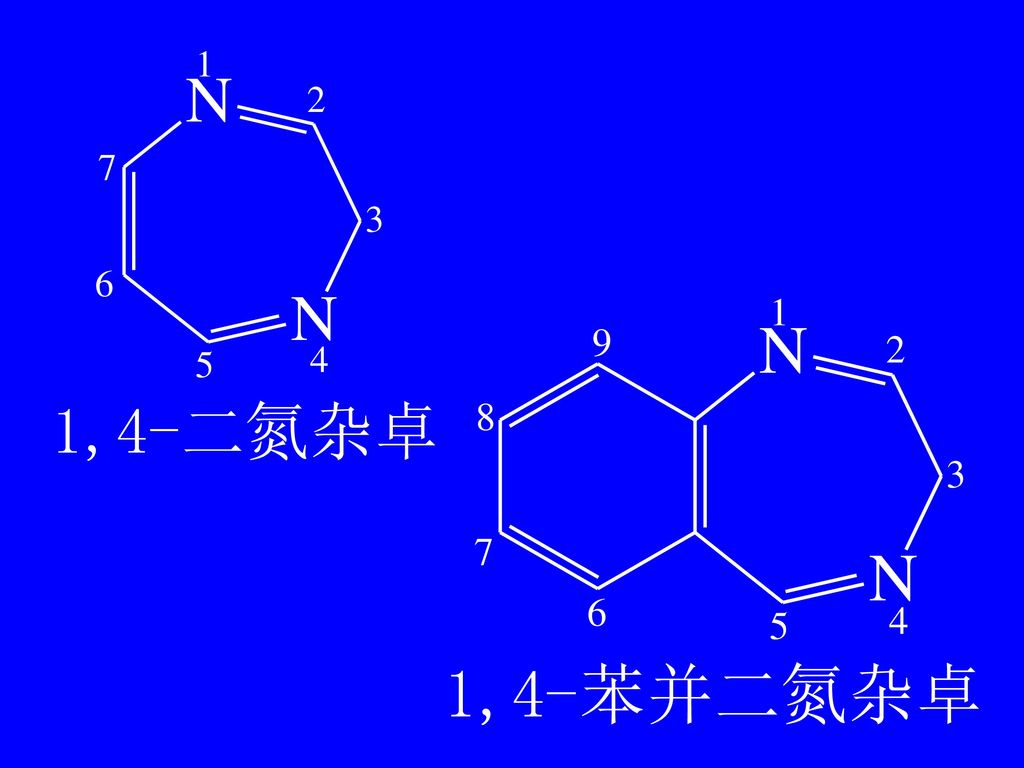
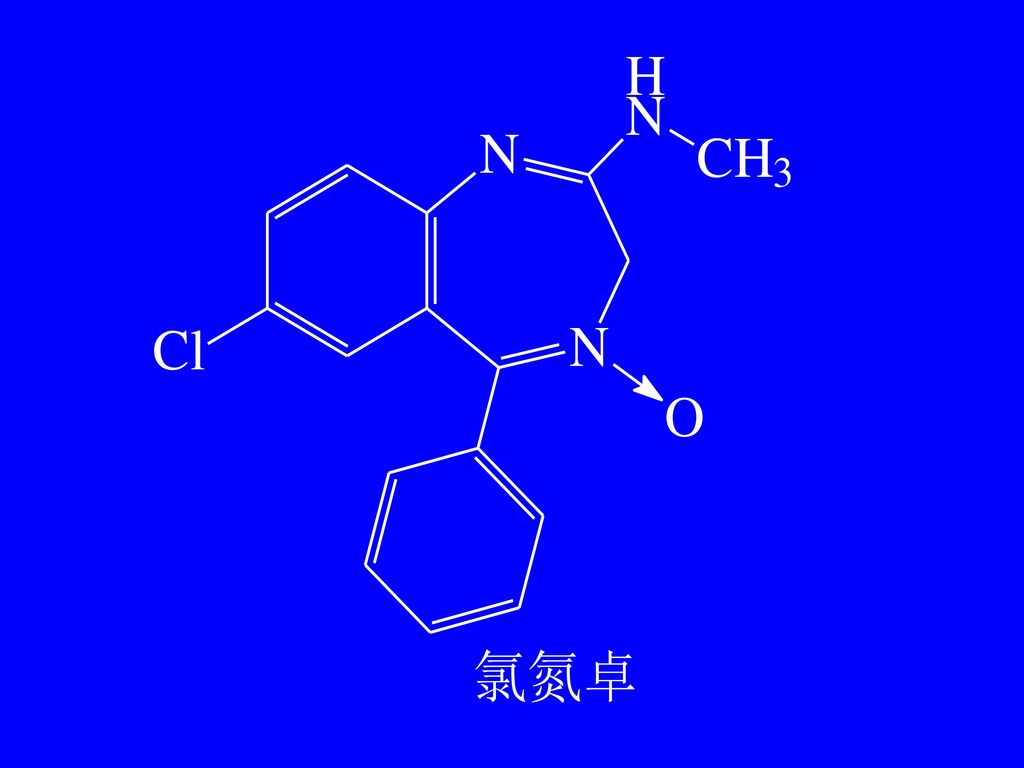
第五节 苯并二氮杂卓 类药物的分析 草 卓 环庚三烯

104
硝西泮

105
氯硝西泮

106
奥沙西泮

107
盐酸氟西泮

108
艾司唑仑

109
阿普唑仑

110
一、结构与性质 1. 苯并二氮杂卓母核 弱碱性,UV 2. 结构中的环一般比较稳定,但在酸性溶液中可水解,形成相应的二苯甲酮衍生物。

112
3. 含卤素

113
二、鉴别试验 (一)化学反应 1. 沉淀反应
化学反应 1. 沉淀反应")
114
氯氮卓 ChP(2000) 【鉴别】(2)取本品约 10mg,加盐酸溶液(9→1000)10ml 溶解后,加碘化铋钾试液 1 滴,即生成橙红色沉淀。
 【鉴别】(2)取本品约 10mg,加盐酸溶液(9→1000)10ml 溶解后,加碘化铋钾试液 1 滴,即生成橙红色沉淀。")
115
阿普唑仑 ChP(2000) [鉴别] (1)取本品约 5mg,加盐酸溶液(9→1000)2ml 溶解后,分为两份:一份加硅钨酸试液 1 滴,即生成白色沉淀;另一份加碘化铋钾试液 1 滴,即生成橙红色沉淀。
![阿普唑仑 ChP(2000) [鉴别] (1)取本品约 5mg,加盐酸溶液(9→1000)2ml 溶解后,分为两份:一份加硅钨酸试液 1 滴,即生成白色沉淀;另一份加碘化铋钾试液 1 滴,即生成橙红色沉淀。](http://slidesplayer.com/slide/11456523/61/images/115/%E9%98%BF%E6%99%AE%E5%94%91%E4%BB%91+ChP%EF%BC%882000%EF%BC%89+%5B%E9%89%B4%E5%88%AB%5D+%EF%BC%881%EF%BC%89%E5%8F%96%E6%9C%AC%E5%93%81%E7%BA%A6+5mg%EF%BC%8C%E5%8A%A0%E7%9B%90%E9%85%B8%E6%BA%B6%E6%B6%B2%EF%BC%889%E2%86%921000%EF%BC%892ml+%E6%BA%B6%E8%A7%A3%E5%90%8E%EF%BC%8C%E5%88%86%E4%B8%BA%E4%B8%A4%E4%BB%BD%EF%BC%9A%E4%B8%80%E4%BB%BD%E5%8A%A0%E7%A1%85%E9%92%A8%E9%85%B8%E8%AF%95%E6%B6%B2+1+%E6%BB%B4%EF%BC%8C%E5%8D%B3%E7%94%9F%E6%88%90%E7%99%BD%E8%89%B2%E6%B2%89%E6%B7%80%EF%BC%9B%E5%8F%A6%E4%B8%80%E4%BB%BD%E5%8A%A0%E7%A2%98%E5%8C%96%E9%93%8B%E9%92%BE%E8%AF%95%E6%B6%B2+1+%E6%BB%B4%EF%BC%8C%E5%8D%B3%E7%94%9F%E6%88%90%E6%A9%99%E7%BA%A2%E8%89%B2%E6%B2%89%E6%B7%80%E3%80%82.jpg "阿普唑仑 ChP(2000) [鉴别] (1)取本品约 5mg,加盐酸溶液(9→1000)2ml 溶解后,分为两份:一份加硅钨酸试液 1 滴,即生成白色沉淀;另一份加碘化铋钾试液 1 滴,即生成橙红色沉淀。")
116
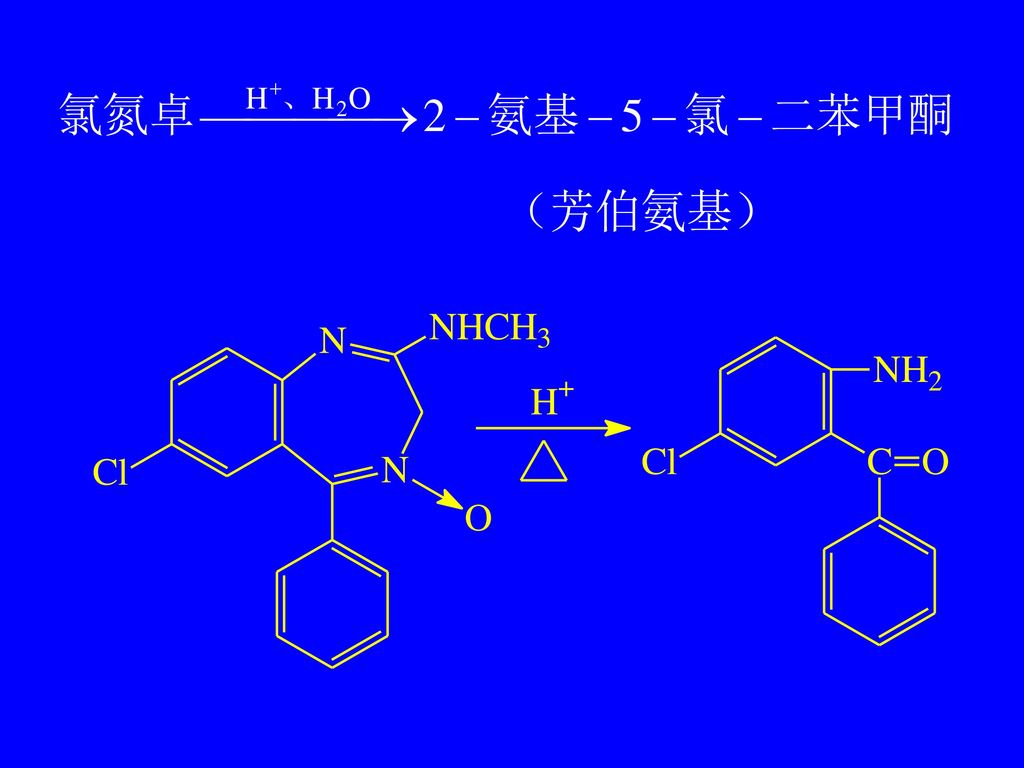
2. 水解后重氮化—偶合反应 氯氮卓、艾司唑仑、奥沙西泮 氯氮卓 ChP(2000) [鉴别] (1)取本品约 10mg,加盐酸溶液(1→2)15ml,缓缓煮沸 15 分钟,放冷,溶液显芳香第一胺类的鉴别反应(附录Ⅲ)。
 [鉴别] (1)取本品约 10mg,加盐酸溶液(1→2)15ml,缓缓煮沸 15 分钟,放冷,溶液显芳香第一胺类的鉴别反应(附录Ⅲ)。 .")
117
3. 水解后呈茚三酮反应 地西泮 4. 硫酸—荧光反应 地西泮 ChP(2000) [鉴别] (1)取本品约 10mg,加硫酸3ml,振摇使溶解,在紫外灯(365nm)下检视,显黄绿色荧光。
 [鉴别] (1)取本品约 10mg,加硫酸3ml,振摇使溶解,在紫外灯(365nm)下检视,显黄绿色荧光。 .")
118
氯氮卓——黄色 艾司唑仑——亮绿色 硝西泮——淡蓝色

119
5. 分解产物的反应 地西泮 ChP(2000) [鉴别] (2)取本品 20mg,用氧瓶燃烧法(附录 Ⅶ C)进行有机破坏,以 5% 氢氧化钠溶液 5ml 为吸收液,燃烧完全后,用稀硝酸酸化,并缓缓煮沸 2 分钟,溶液显氯化物的鉴别反应(附录 Ⅲ)。
 [鉴别] (2)取本品 20mg,用氧瓶燃烧法(附录 Ⅶ C)进行有机破坏,以 5% 氢氧化钠溶液 5ml 为吸收液,燃烧完全后,用稀硝酸酸化,并缓缓煮沸 2 分钟,溶液显氯化物的鉴别反应(附录 Ⅲ)。 .")
120
(二)UV 和 IR (三)TLC
UV 和 IR (三)TLC")
121
三、特殊杂质的检查 (一) 有关物质的检查 1. 地西泮中有关物质的检查 地西泮的原料及片剂 TLC 高低浓度对比法
 有关物质的检查 1. 地西泮中有关物质的检查 地西泮的原料及片剂 TLC 高低浓度对比法")
122
地西泮(安定) N O C l H 3
 N O C l H 3")
123
2. 氯氮卓中有关物质的检查 USP(30) TLC 氧化物、氨基物 杂质对照品法 有关物质 参比杂质对照品法
氧化物、氨基物 杂质对照品法 有关物质 参比杂质对照品法 BP(2007)HPLC 高低浓度对比法 A、B、C 杂质对照品法 3. 氯氮卓中有关物质的反相高效液相色谱法
HPLC 高低浓度对比法. A、B、C 杂质对照品法. 3. 氯氮卓中有关物质的反相高效液相色谱法.")
124
氯氮卓(利眠宁) 水解后为芳伯氨基 最不活泼型Cl
 水解后为芳伯氨基 最不活泼型Cl")
125
(二)降解产物的检查 地西泮注射液 分解产生2-甲氨基-5-氯二苯甲酮 HPLC 主成分自身对照法
降解产物的检查 地西泮注射液 分解产生2-甲氨基-5-氯二苯甲酮 HPLC 主成分自身对照法")
126
1. 峰面积归一化法 取一定量供试品溶液进样,测定各杂质及药物的峰面积,计算各杂质峰面积及其总和占总峰面积的百分率,不能超过限量。注意溶剂峰不应计算在总峰面积内。

127
优点:不需杂质对照品,简便易行。 缺点:检测波长处,各杂质和药物的吸收强度可能有差异,控制杂质峰面积百分率不一定就是杂质限量,准确度差。

128
2. 主成分自身对照法 杂质峰面积与主成分峰面积相差悬殊时应用。 方法

129
分别进样,测定供试品溶液中各杂质峰面积及其总和,与对照溶液主成分峰面积比较,控制供试品中杂质的量。

130
优点:不需杂质对照品,可同时控制各个杂质及其总量限度
缺点:检测波长处,各杂质和药物的吸收强度可能有差异,准确度差

131
3. 内标法测定供试品中杂质的总量限度 方法 供试品→供试品溶液1 图1 供试品+内标→供试品溶液2 图2

132
判断 若图1无与内标峰保留时间相同的杂质峰,则图2中各杂质峰面积之和应小于内标物质峰面积(溶剂峰不计在内)
")
133
若图1有与内标峰保留时间相同的杂质峰,则将图2上的内标峰面积减去图1中此杂质峰面积,得内标物质峰的校正面积;图2中各杂质峰面积加上图1此杂质峰面积,得各杂质峰的校正总面积;各杂质峰校正总面积应小于内标物质峰的校正面积。

134
4. 内标法加校正因子测定供试品中某个杂质的含量
准确测定杂质含量 方法 内标+杂质对照品→溶液,进样,测定,计算校正因子

135
供试品+内标→供试品溶液,进样,测定杂质和内标峰面积或峰高,计算杂质量

136
5. 外标法测定供试品中某个杂质的量 供试品→供试品溶液 杂质对照品→对照品溶液进样,测定,计算杂质的含量

137
缺点:不易准确控制进样量,宜用定量环进样

138
四、含量测定 (一)非水溶液滴定法 原料 (二)紫外分光光度法
非水溶液滴定法 原料 (二)紫外分光光度法")
139
(三)比色法 1. 地西泮经水解后氯仿提取液的比色测定法
比色法 1. 地西泮经水解后氯仿提取液的比色测定法")
140
2. 地西泮水解产物甘氨酸与茚三酮反应后比色测定法

141
3. 氯氮卓经酸水解后的重氮化—偶合比色测定法

142
(四)HPLC 1. 地西泮注射液的 HPLC 法 内标法加校正因子定量 2. 三唑仑的 HPLC 法
HPLC 1. 地西泮注射液的 HPLC 法 内标法加校正因子定量 2. 三唑仑的 HPLC 法")
Similar presentations















 淮海工学院 制药工程系 马卫兴>")





