Download presentation
Presentation is loading. Please wait.

1
基因的分离与鉴定

2
DNA克隆片段的产生和分离 重组体DNA分子的构建及导入受 体细胞 基因克隆的实验方案 克隆基因的分离 重组子的选择与鉴定

3
DNA克隆片段的产生和分离 1. 利用限制性内切酶片段化基因组DNA
鸟枪法(shotgun approch):用限制性内切酶消化给体基因组DNA,这种消化产物,不经过凝胶电泳分部分离,就直接用来同载体分子作连接反应的克隆方法。 2. 凝胶电泳法和蔗糖梯度离心法分离
:用限制性内切酶消化给体基因组DNA,这种消化产物,不经过凝胶电泳分部分离,就直接用来同载体分子作连接反应的克隆方法。 2. 凝胶电泳法和蔗糖梯度离心法分离.")
4
构 建 重 组 DNA 分 子 的 途 径

5
重组子分子导入受体细胞的途径 原核生物的转化与转染 真核生物的转染

6
原核生物的转化与转染 E.coli的钙转化 E.coli的电穿孔转化 转染 转导

7
E.coli的钙转化 细菌细胞在一定的生理状态(感受态),或经CaCl2处理,或制备成原生质体的情况下,都可吸收外源DNA,利用这一特性可将重组DNA导入受体细胞中,用质粒作载体的遗传工程一般使用这种方法。 由于E.coli不会自发地产生感受态,因此E.coli的转化一般采用钙转化法。该方法是将E.coli用冷CaCl2(0℃) 致敏,而后在高温(42 ℃)下热休克,这样可使外原DNA进入受体细胞。
 致敏,而后在高温(42 ℃)下热休克,这样可使外原DNA进入受体细胞。")
8
E.coli的电穿孔转化 在高压电场下使细胞产生瞬间的穿孔,这个穿孔足够大并可维持足够的时间,使外原DNA进入受体细胞。电穿孔法的转化效率比钙转化法高2~3个数量级。这种方法也可用于真核生物的转染。

9
转染是指转化感染(Transfection来自于transfomation与infection)
凡是以噬菌体(如M13)或病毒为载体,以转化的方法将DNA导入细胞的方法均称为转染。因此转染就方法来说与转化是一样的。
或病毒为载体,以转化的方法将DNA导入细胞的方法均称为转染。因此转染就方法来说与转化是一样的。")
10
体外包装颗粒的转导 将重组的 λ噬菌体DNA或重组的柯斯载体DNA经体外包装成具有感染能体力的λ噬菌体颗粒,然后经由在受体细胞表面上的λDNA接受位点,使这些带有目的基因序列的重组体DNA注入大肠杆菌。

11
真核生物的转染 1. 磷酸钙转染技术 2.电穿孔法 3.基因抢转染法 4.激光微束穿孔法 5.显微注射法 6.脂质体介导法 7.多聚物介导法

12
磷酸钙转染技术 主要用于动物细胞的转染。将DNA溶液加入到CaCl2中,然后在强烈震荡下加入由Na2HPO4和NaH2PO4组成的磷酸缓冲液,使溶液产生磷酸钙沉淀并将DNA包裹在沉淀中。将这些沉淀加入到细胞中,利用细胞的胞饮作用将DNA导入到细胞中。磷酸钙一方面可以增加细胞的通透性,一方面可以避免DNA被细胞内的核酸酶水解。

13
电穿孔法:原理与原核生物的电穿孔法一样,主要用于植物细胞的转染。
基因抢转染法:主要用于植物细胞的转染。利用被电场或机械加速的金属微粒能够进入细胞内的基本原理,先将DNA溶液与钨、金等金属微粒一起保温,使DNA吸附在金属微粒表面,随高速的金属微粒直接进入细胞内。

14
激光微束穿孔法:主要用于叶绿体或线粒体的基因工程操作。利用直径很小、能量很高的激光束在细胞表面引起可逆性的穿孔,使DNA进入细胞。

15
脂质体(liposome)介导法:主要用于动物细胞或植物的原生质体。将DNA包裹在人工制备的磷脂双分子层的膜状结构内,通过脂质体与细胞膜的融合而将DNA导入细胞内。
多聚物介导法:用于动物细胞或植物的原生质体。利用多聚物如PEG、二乙胺乙基葡聚糖(DEAE葡聚糖)、多聚赖氨酸、多聚鸟氨酸等与二价阳离子、DNA混合形成沉淀颗粒,通过细胞的胞饮作用而进入细胞内。
、多聚赖氨酸、多聚鸟氨酸等与二价阳离子、DNA混合形成沉淀颗粒,通过细胞的胞饮作用而进入细胞内。")
16
基因克隆的实验方案 1. cDNA基因克隆 2. 基因组DNA克隆 3. 基因定位克隆

17
cDNA基因克隆 cDNA libraries

18
cDNA基因克隆 cDNA文库是指得到足够多的分别克隆的cDNA片段,汇集这些克隆应包含某种细胞中各种mRNA的相应顺序,每种顺序至少有一份拷贝,这种克隆片段的汇集体称为某种细胞的cDNA文库。

19
cDNA libraries mRNA isolation, purification Check theRNA integrity
Fractionate and enrich mRNA Synthesis of cDNA Treatment of cDNA ends Ligation to vector

20
cDNA文库 没有原核生物的cDNA 文库 原核生物的 mRNA 非常不稳定; 原核生物的基因组文库比较容易构建,能包含所有基因的序列。

21
cDNA 文库 cDNA 文库对于真核生物的基因分析 cDNA文库是只含有编码蛋白的基因文库 cDNAs 没有内含子 基因可以在E. coli 中直接表达 用于新基因的鉴别 组织或特殊类型细胞(基因的特异表达)
 .")
22
大部分真核生物的 mRNAs 有 3’ poly( A )尾巴
cDNA 文库-mRNA 分离 大部分真核生物的 mRNAs 有 3’ poly( A )尾巴 oligo (dT) 能与poly(A) 尾巴互补,由此来获得mRNA. AAAAAAAAAAn 5’ cap
尾巴. oligo (dT) 能与poly(A) 尾巴互补,由此来获得mRNA. AAAAAAAAAAn. 5’ cap.")
24
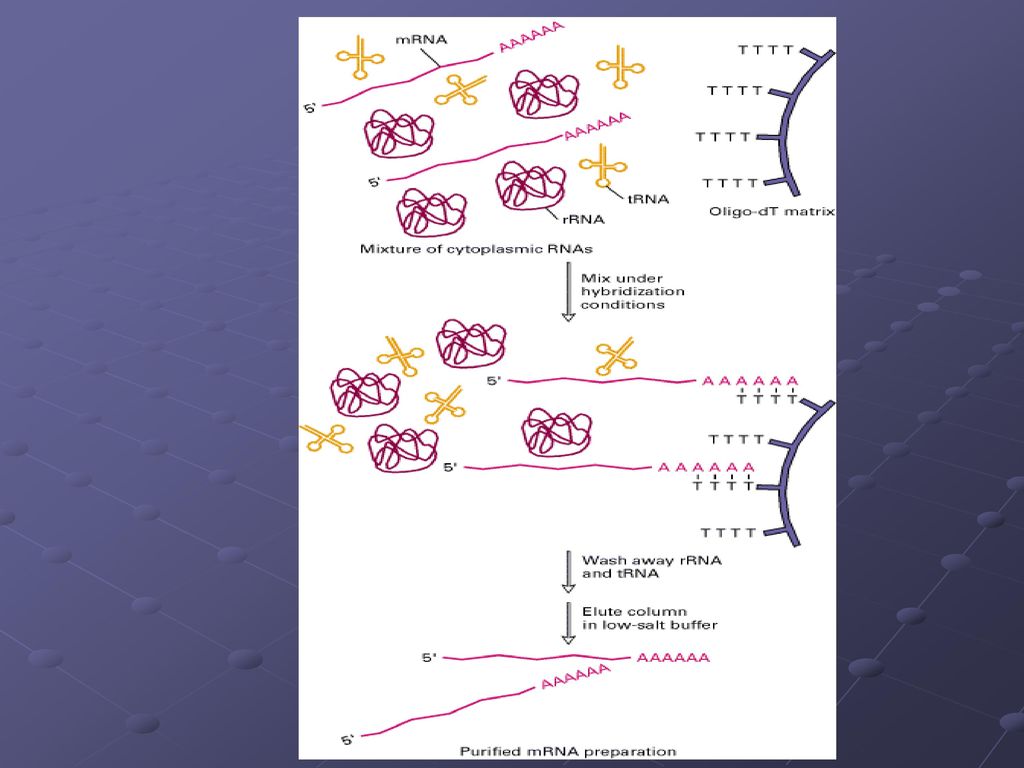
三种分离mRNA的方法 1. 传统的分离方法是用oligo (dT)-纤维素柱 分离总RNA
2. 把oligo(dT) -磁珠同总RNA混合,在强磁场下分离mRNA 3.用蔗糖梯度离心mRNA核糖体复合体(溶解细胞)来分离mRNA
 -磁珠同总RNA混合,在强磁场下分离mRNA. 3.用蔗糖梯度离心mRNA核糖体复合体(溶解细胞)来分离mRNA.")
25
用纤维素纯化poly(A)mRNA的流程图
mRNA的流程图")
26
cDNA 文库- 检测mRNA的完整性 确定 mRNA 没被降解 方法: mRNA的翻译 : 用体外蛋白质合成检测mRNAs 。(麦胚无细胞转译体系或兔网织细胞裂解物转译体系) 通过凝胶电泳分析mRNAs : 用琼脂糖或聚丙烯酰胺凝胶电泳
 通过凝胶电泳分析mRNAs : 用琼脂糖或聚丙烯酰胺凝胶电泳.")
27
克隆特殊的 mRNAs 凝胶分离: 通过琼脂糖凝胶电泳回收 富集: 通过杂交来实现 mRNAs(根据mRNA的大小)
克隆一个特殊的基因比建立完整的cDNA文库更有用 凝胶分离: 通过琼脂糖凝胶电泳回收 mRNAs(根据mRNA的大小) 富集: 通过杂交来实现
 富集: 通过杂交来实现.")
28
cDNA 的合成: 第一链 的合成: mRNA, 反转录酶,oligo(dT) , 第二链的合成: 根据在第一链3’-末端加尾
核酸末端转移酶,dNTPs 1. oligo(dT) 引导的cDNA合成法 2. 随机引物引导的cDNA合成法 第二链的合成: 根据在第一链3’-末端加尾 (C),用补充引物合成第二链
 引导的cDNA合成法. 2. 随机引物引导的cDNA合成法. 第二链的合成: 根据在第一链3’-末端加尾. (C),用补充引物合成第二链.")
29
The first strand synthesis
mRNA 5’ AAAAA-3’ HO-TTTTTP-5’ Reverse transcriptase Four dNTPs mRNA 5’ AAAAA-3’ 3’ TTTTTP-5’ cDNA Terminal transferase dCTP mRNA 5’ AAAAACCC-3’ 3’-CCCCCCC TTTTTP-5’ cDNA Alkali (hydrolyaes RNA) Purify DNA oligo(dG) 5’-pGGGG-OH 3’-CCCCCCC TTTTTP-5’ cDNA Klenow polymerase or reverse Transcriotase Four dNTPs 5’-pGGGG -3’ 3’-CCCCCCC TTTTTP-5’ Duplex cDNA The first strand synthesis
 Purify DNA oligo(dG) 5’-pGGGG-OH. 3’-CCCCCCC. TTTTTP-5’ cDNA. Klenow polymerase or reverse. Transcriotase Four dNTPs. 5’-pGGGG. -3’ 3’-CCCCCCC. TTTTTP-5’ Duplex cDNA. The first strand synthesis.")
30
Single strand-specific nuclease
Duplex cDNA 5’-pGGGG -3’ 3’-CCCCCCC TTTTTp-5’ Single strand-specific nuclease 5’-pGGGG -3’ 3’-CCCC TTTTTp-5’ Klenow polymerase treat with E.coRI methylase 5’-pGGGG -3’ 3’-CCCC TTTTTp-5’ Add E.colRI linkers using T4 DNA ligase HO-CCG/AATTCGG-3’ 3’-GGCTTAA/GCC-OH HO-CCGAATTCGGGGGG CCGAATTCGG-3’ 3’-GGCTTAAGCCCCCC TTTTTGGCTTAAGCC-OH E.colRI digestion 5’-pAATTCGGGGGG CCG-3’ 3’-CCCCCCC TTTTTGGCTTAAp-5’ Ligate to vector and transfom Second strand synthesis

31
对cDNA 末端的处理 平末端的片断需要加入特殊的核苷酸序列形成粘性末端,才能进行克隆 步骤 :
移去突出的 3’-ends(strand-special nuclease) 填充缺失 3’ 核苷酸 (klenow fragment of DNA polyI and 4 dNTPs) 衔接物和平末端的连接(T4 DNA ligase) 限制性内切酶的消化 (E.coRI )
 填充缺失 3’ 核苷酸 (klenow fragment of. DNA polyI and 4 dNTPs) 衔接物和平末端的连接(T4 DNA ligase) 限制性内切酶的消化 (E.coRI )")
32
与载体的连接 载体末端脱磷酸(碱性磷酸酶) 用T4 DNA 连接酶连接 载体 和 cDNA 步骤 :
任何一个含有E.coRI 酶切位点的载体都可以用来克隆 cDNA. 步骤 : 载体末端脱磷酸(碱性磷酸酶) 用T4 DNA 连接酶连接 载体 和 cDNA (plasmid or l phage vector)
 用T4 DNA 连接酶连接 载体 和 cDNA. (plasmid or l phage vector)")
33
(一些RNA病毒,不经过DNA中间体,对其研
cDNA克隆的优越性 1. 以mRNA为材料; (一些RNA病毒,不经过DNA中间体,对其研 究,cDNA是唯一可行的方法) 2. 基因文库的筛选比较简单易行; 3. 由于每一个cDNA克隆都含有一种mRNA序列, 在选择中出现假阳性的几率比较低; 4. 克隆在细菌中表达的基因,用作基因序列的测定, 和发育过程中或组织中基因特异表达
 2. 基因文库的筛选比较简单易行; 3. 由于每一个cDNA克隆都含有一种mRNA序列, 在选择中出现假阳性的几率比较低; 4. 克隆在细菌中表达的基因,用作基因序列的测定, 和发育过程中或组织中基因特异表达.")
34
基因组DNA克隆 Genomic libraries

35
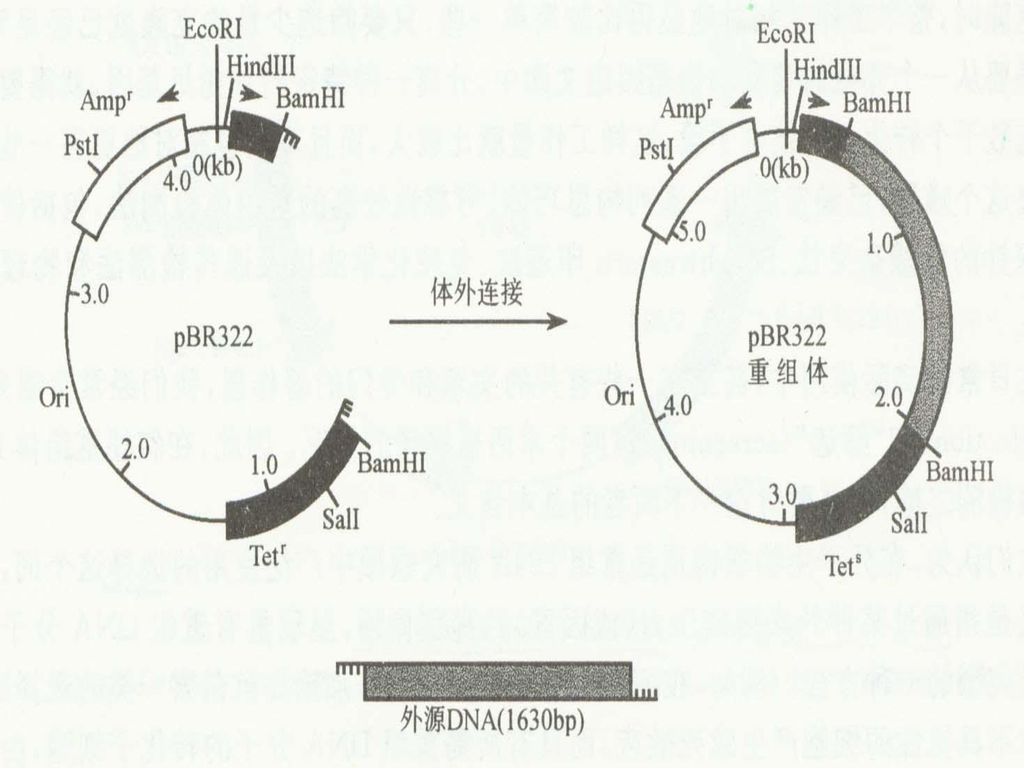
基因组文库的构建 基因文库(gene library):用重组DNA技术将某种生物细胞的总DNA或染色体DNA的所有片段随机地连接在载体上,然后转移到适当的宿主细胞中通过细胞增殖而构成各个片段的克隆。当制备的克隆数目多到可以把某种生物的全部基因都包含在内的情况下,这一组克隆的总体就称为某种生物的基因文库。同一定义也适用于线粒体或叶绿体DNA的基因文库。由于制备DNA片段的切点是随机的,所以每一克隆内所含的DNA片段即可能是一个或几个基因,也可能是一个基因的一部分或除完整基因外还包含着两侧的邻近DNA顺序。
:用重组DNA技术将某种生物细胞的总DNA或染色体DNA的所有片段随机地连接在载体上,然后转移到适当的宿主细胞中通过细胞增殖而构成各个片段的克隆。当制备的克隆数目多到可以把某种生物的全部基因都包含在内的情况下,这一组克隆的总体就称为某种生物的基因文库。同一定义也适用于线粒体或叶绿体DNA的基因文库。由于制备DNA片段的切点是随机的,所以每一克隆内所含的DNA片段即可能是一个或几个基因,也可能是一个基因的一部分或除完整基因外还包含着两侧的邻近DNA顺序。")
36
基因文库的构建就是利用所谓的“鸟抢法”,使基因组的DNA片段随机地插入适当的载体中,引入细胞进行大量繁殖而构建的。

37
基因组 DNA 文库 eukaryotes 纯化基因组DNA prokaryotes 基因组DNA的片断化 物理切割和限制性内切酶
与载体连接

38
基因组 DNA的纯化 : 真核生物 :从细胞核中 原核生物:直接从细胞中体取基因组DNA
去除蛋白(蛋白酶消化), 脂类和其它“杂质”。 原核生物:直接从细胞中体取基因组DNA
, 脂类和其它 杂质 。 原核生物:直接从细胞中体取基因组DNA.")
39
限制性内切酶消化 将片断的末端 (粘性或平头) 和酶消化的载体连接
Hae :5’- …GG /CC… 3’, Sau3A: 5’-/GATC-3’, Alu: 5’- …AG/CT… 3’, BamH1: 5’-G/GATCC 酶切位点是否被修饰( 在哺乳动物种CpG 甲基化) 内切酶消化的时间和用量取决于要插入片断的大小范围。
 内切酶消化的时间和用量取决于要插入片断的大小范围。")
40
用于构建基因文库片段的产生大致有两种方法:
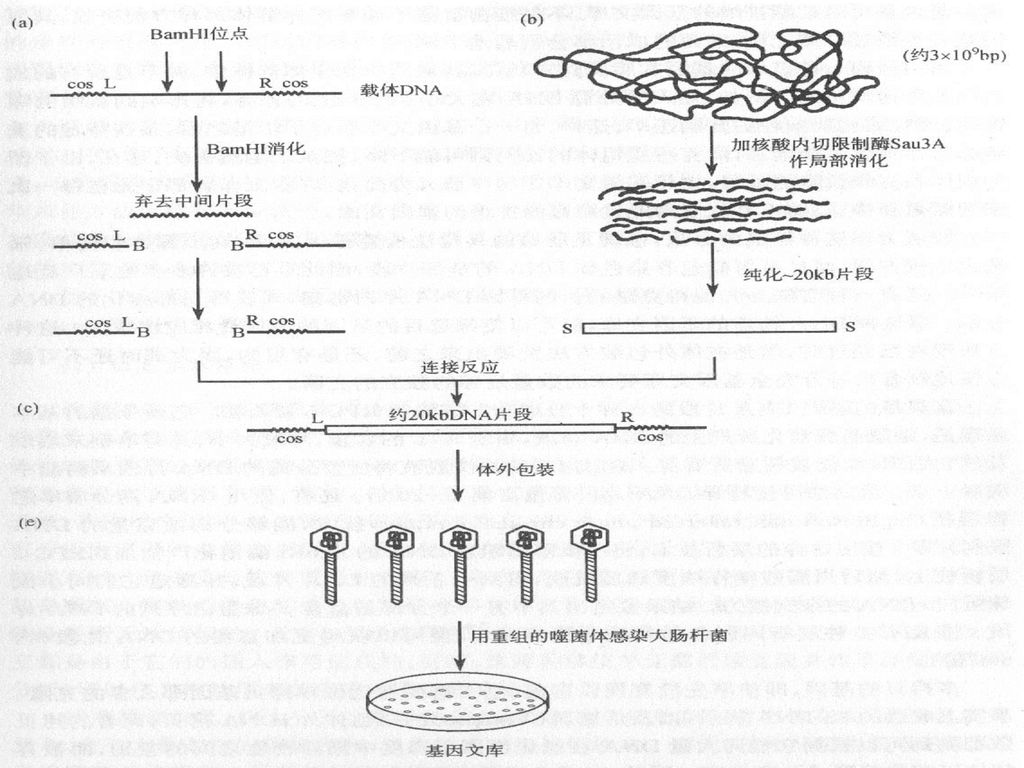
一是用限制性内切酶完全消化基因组DNA,这个方法有两个特点。第一是假如所需的基因含有所用限制酶的识别位点,那么这一基因将被克隆在两个或数个片段中,给研究带来一定的困难。第二,一般常用的限制酶大多识别六个bp序列,切割DNA所产生的片段的平均大小相对比较小(约4kb即46=4096bp),因此整个基因文库要含有非常大量的重组体,这样应用分子杂交法进行筛选就十分费事费时。虽然可以用限制酶进行部分消化,产生约20kb的片段,但这种方法必须掌握好酶解的条件。
,因此整个基因文库要含有非常大量的重组体,这样应用分子杂交法进行筛选就十分费事费时。虽然可以用限制酶进行部分消化,产生约20kb的片段,但这种方法必须掌握好酶解的条件。")
41
用于制备基因文库的随机片段的另一种方法是采用机械随机切割。这种方法可以制备大片段的DNA(用噬菌体λ作载体约20kb,以cosmid作载体约45kb ),从而克服上述的两处缺点。但是这种方法的不是之处是:所制备的片段的末端顺序是随机的,还必须经过末端修复,人工制造接头等手续才能与载体DNA进行连接。 用于基因文库构建的载体常用噬菌体λ或cosmid载体,因为它们的容量比较大,可克隆大片段的DNA。虽然cosmid载体比λ载体的容量大,但由于文库的保存(cosmid载体在E.coli中, λ载体在噬菌体中) λ比cosmid容易,因此λ载体用的更多些。
,从而克服上述的两处缺点。但是这种方法的不是之处是:所制备的片段的末端顺序是随机的,还必须经过末端修复,人工制造接头等手续才能与载体DNA进行连接。 用于基因文库构建的载体常用噬菌体λ或cosmid载体,因为它们的容量比较大,可克隆大片段的DNA。虽然cosmid载体比λ载体的容量大,但由于文库的保存(cosmid载体在E.coli中, λ载体在噬菌体中) λ比cosmid容易,因此λ载体用的更多些。")
42
Hae 、 Alu

44
文库的大小 (确定有足够的克隆数) 必须包括一定数量的重组子才可能克隆基因组中的任何碱基序列。 计算重组子数量的公式: ln (1-P)
ln (1-f) N:一个完整基因文库所应包含的重组DNA的转化子的克隆数 P: 重组体群体中出现目的基因的序列的几率(一般期望99% ) f : 限制片断的平均大小与基因组DNA总量之比
 N:一个完整基因文库所应包含的重组DNA的转化子的克隆数. P: 重组体群体中出现目的基因的序列的几率(一般期望99% ) f : 限制片断的平均大小与基因组DNA总量之比.")
45
For example : 期望值为0.99,插入片断为20kb的 E.coli (4.6×106 bp) 和 human (3×109 bp) 基因组的克隆数的计算 N E.coli= =1.1 ×103 ln( ) ln[1-(2×104/4.6×106)] ln(1-0.99) Nhuman= = ×105 ln[1-(2 ×104/3 ×109)] 这个例子说明用质粒载体(插入片断5-10kb )就可以建立很好的原核生物基因文库,只需要几千个克隆;而真核生物则需要更大承载能力的载体。
 ln[1-(2×104/4.6×106)] ln(1-0.99) Nhuman= = 6.9 ×105. ln[1-(2 ×104/3 ×109)] 这个例子说明用质粒载体(插入片断5-10kb )就可以建立很好的原核生物基因文库,只需要几千个克隆;而真核生物则需要更大承载能力的载体。")
46
2.目的基因包容在一个其大小范围超出载体承载能力的DNA片断上
有一些序列不能被克隆 Example: 1. 序列缺乏酶切位点; 2.目的基因包容在一个其大小范围超出载体承载能力的DNA片断上 Too long for the vector used

47
载体 根据基因组的大小选择合适的载体构建DNA文库。 载 体 Plasmid phageλ cosmid BAC YAC
插入片断 (kb) 最常用的载体是 phageλ
 最常用的载体是. phageλ.")
48
基因定位克隆 Map-baseed cloning

49
主要内容: 1. 拟南芥简介 2. RFLP分子标记 3. RFLP作图的原理与步骤 4. 染色体步移 5. 大尺度基因组物理图谱的构建

50
基因定位克隆 是用于分离其编码产物上不知道的目的基因的一种有效的方法。从理论上讲,任何一种可鉴定出有一个突变的基因,都可以通过基因定位克隆技术予以分离。

51
1.先将目的基因定位到染色体上,并在目的基因的两侧确定一对紧密连接的RFLP或RAPD分子标记;
步骤: 1.先将目的基因定位到染色体上,并在目的基因的两侧确定一对紧密连接的RFLP或RAPD分子标记; 2. 利用最紧密连锁的一对两侧分子标记作探针,通过染色体步移技术将位于这两个分子标记之间的含目的基因组片断克隆并分离出来; 3. 最后根据其同突变体发生遗传互补的能力从此克隆重鉴定出目的基因。

52
应用基因定位克隆技术 分离目的基因的必要条件: 2.有可用的同目的基因紧密连锁的DNA探针,(距离几百bp )
1. 以酵母人工染色体为载体构建含有大片断DNA的YAC文库。 2.有可用的同目的基因紧密连锁的DNA探针,(距离几百bp )
")
53
拟南芥简介: 1. 植株小,便于筛选突变体; 2. 时代时间短(5个星期左右),可以积累大量遗传数据);
3. 种子产率高,每株可产生4104粒以上的种子; 4. 基因组小,结构简单,总长度7 107bp适合于用染色体步移技术克隆目的基因; 5. 天然自花授粉,可得到突变基因的纯合子,同时也可通过人工杂交授粉,以便给基因定位。

54
DNA限制片断长度多态性,它是指应用特定的核酸内切酶切割有关的DNA分子,所产生的DNA片断在长度上的简单变化。
RFLP 分子标记 DNA限制片断长度多态性,它是指应用特定的核酸内切酶切割有关的DNA分子,所产生的DNA片断在长度上的简单变化。 由于核酸内切酶是以一种序列特异的方式切割DNA分子,来自一个完整的纯合子个体(所有的基因及DNA序列)的每一种同源DNA分子,都会在同样的位点被准确地切割。
的每一种同源DNA分子,都会在同样的位点被准确地切割。")
55
它所代表的是基因组DNA在限制性内切酶消化后产生片段在长度上差异,由于不同个体等位基因之间碱基的互换、重排、缺失等变化导致限制内切酶识别位点发生改变,从而造成基因型间限制性片段长度的差异。

56
当这些大小不等的片段通过凝胶电泳时,就形成不同带,通过与克隆的DNA探针进行Southren杂交和放射自显影后,即获得反映个体特异性的RFLP图谱。

57
生态型A的DNA 基因A 生态型B的DNA 基因A EcoR EcoR EcoR EcoR分别切割两种生态型DNA
GAATTC GAATTC GAATTC CTTAAG CTTAAG CTTAAG EcoR EcoR EcoR 生态型B的DNA 基因A GAATTC GGATTC GAATTC CTTAAG CCTAAG CTTAAG EcoR分别切割两种生态型DNA 电泳后作Southen杂交 同放射性同位素标记的基因A克隆杂交 a b

58
RFLP作图的原理和步骤: C N a b X 探针A 探针B 探针B 探针A H F1杂合子 探针A 探针B F2代

59
A 2 : 1 : 1 B H C N 自由组合 不连锁 紧密连锁 需要筛选相当大量的F2代群体, 通过重组子计算基因A与B之间
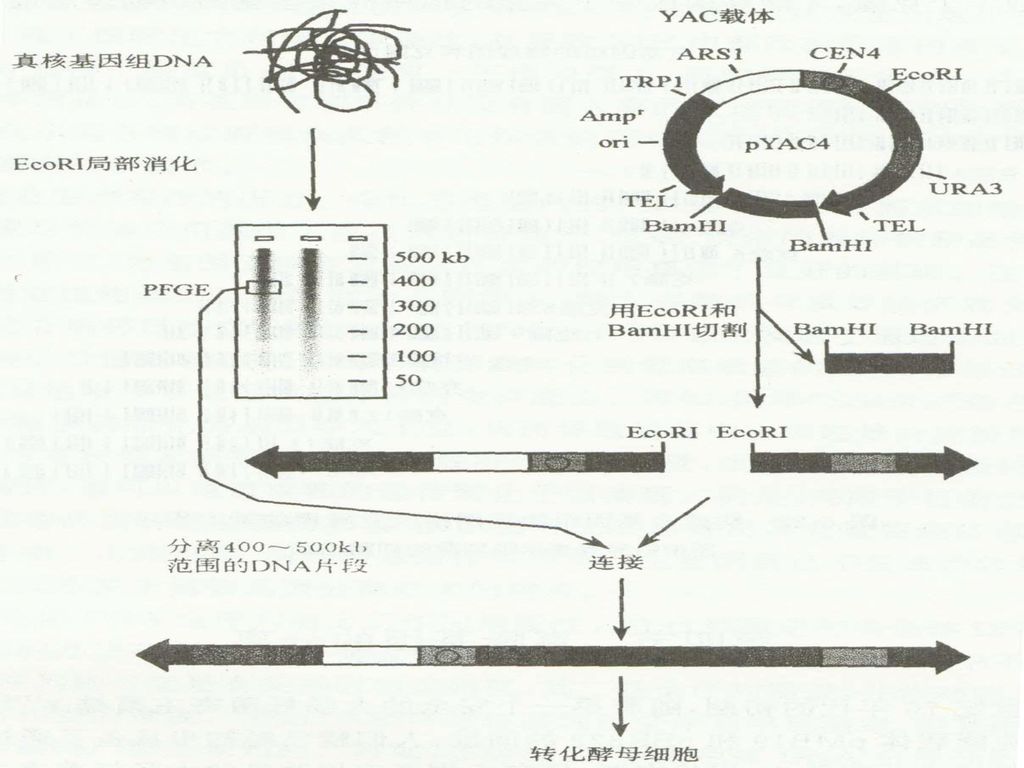
2 : 1 : 1 B 自由组合 H C N 需要筛选相当大量的F2代群体, 通过重组子计算基因A与B之间 的遗传距离 。

60
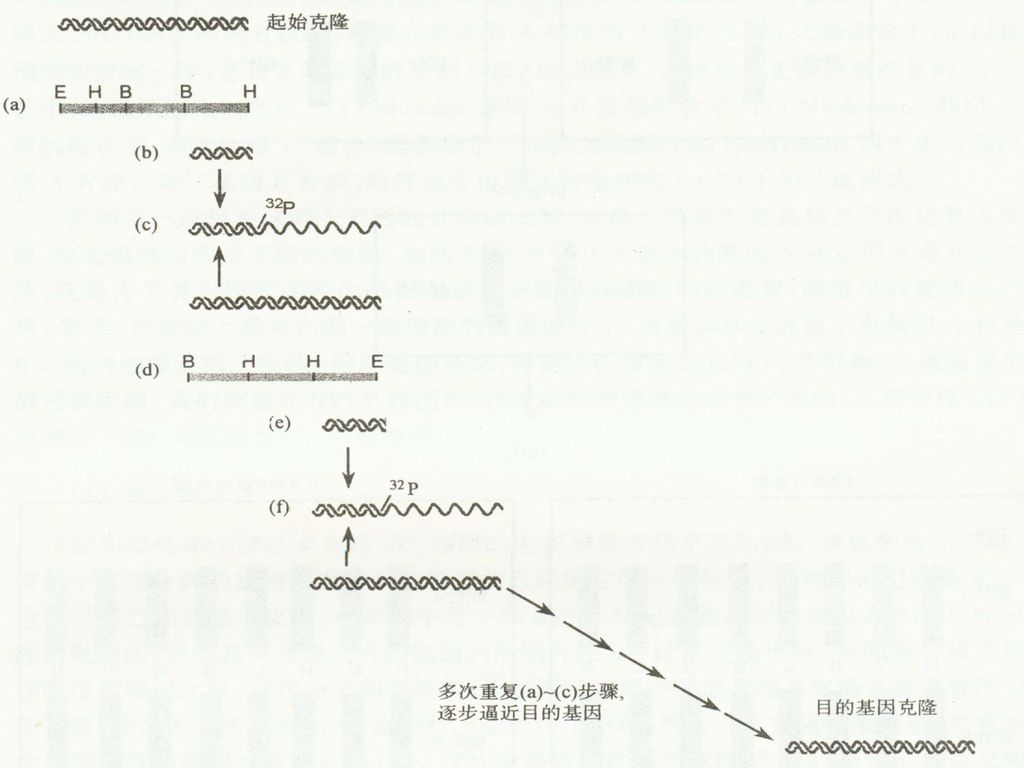
染色体步移(chrosome walking)
这种技术通过逐一克隆来自染色体基因组DNA的彼此重叠的序列,而慢慢靠近目的基因,因此形象的称为染色体步移。 步骤 1. 先构建起点RFLP标记克隆的限制图,把其中最靠近目的基因的限制片断亚克隆出来; 2. 经同位素标记之后,用作分子杂交探针; 从基因组文库中筛选与起点克隆具有重叠序列的新克隆; 重复进行上述步骤,直到找到目的基因的克隆。 。

62
染色体步移的影响因素: 1. 在基因组的不同位置上散布着重复的DNA序列,它们会扰乱步移的顺序性,因此,进行染色体步移的探针必须是唯一的序列的克隆。 2. 在基因组DNA中存在着一些无法克隆的序列,因为当它们被克隆到所使用的克隆载体上时,会发生致死效应。

63
在拟南芥和番茄中1cM的图距大约相当于290 kb和750 kb,而在人类基因组中1cM的图距大约相当于1 000 kb,在小麦中这个数字是 kb。 由于大多数高等真核基因组中都存在着大量的散在重复序列,致使染色体步移对于超大型的DNA序列的作图和超长的距离进行基因定位克隆存在一定的困难。

64
大尺度基因组物理图谱的构建 1. DNA分子大片断切割 2. 大片断DNA分子的分离 3. YAC库的构建 4. 大尺度物理图谱的构建

65
DNA分子大片断切割 用切割位点稀少的核酸内切酶进行切割。 大片断DNA分子的分离 脉冲电场凝胶电泳(PFGE)
")
66
YAC库的构建 这类载体含有 1. 酵母菌的复制起点ARS(automatic replication sequence,ARS),
YAC(yeast artificial chromosome,酵母人工染色体)质粒载体是在人类基因组计划(HGP)实施的背景下发展起来的一类克隆特大片段的载体。 这类载体含有 1. 酵母菌的复制起点ARS(automatic replication sequence,ARS), 2. 来自酵母染色体的着丝粒CEN(centromere)序列; 3. 一对酵母的端粒TEL(telomere)序列;
质粒载体是在人类基因组计划(HGP)实施的背景下发展起来的一类克隆特大片段的载体。 这类载体含有. 1. 酵母菌的复制起点ARS(automatic replication sequence,ARS), 2. 来自酵母染色体的着丝粒CEN(centromere)序列; 3. 一对酵母的端粒TEL(telomere)序列;")
67
4. 选择性标记和克隆位点; 5. 同时还含有大肠杆菌的复制起始点,可以在大肠杆菌中以环状质粒的形式存在。 DNA体外重组后以原生质体转化的方式导入酵母细胞。 这类载体克隆能力可达1~2Mb。主要用于基因组文库的构建。

69
大尺度物理图谱的构建 物理图谱(physical genes):分子标记间的物理图距是以核苷酸数目表示的,这种图谱现在可以PFGE法进行构建。通过测定携带分子标记的DNA限制片断的大小,便可以计算出两个分子标记之间的物理图距。
:分子标记间的物理图距是以核苷酸数目表示的,这种图谱现在可以PFGE法进行构建。通过测定携带分子标记的DNA限制片断的大小,便可以计算出两个分子标记之间的物理图距。")
70
构建全基因组物理图谱的过程 分离到大量的基因组克隆,并构建它们的详细的限制图谱,就可以鉴定出重叠的基因组克隆,进而构建出全基因组的物理图谱。

72
克隆基因的分离 1.应用核酸探针 2.应用差别杂交或扣除杂交法 3.应用mRNA差别显示技术 4.应用表达文库 5.酵母双杂交体系

73
应用核酸探针 分离克隆的目的基因

74
步骤: 1.获得探针 2.转膜 3.杂交

75
antibody or enzyme Transfer the DNA in the plaque or colony to a
Nylon or nitrocellulose membrane Phage DNA bind to the membrane directly Bacterial colonies must be lysed to release DNA on the membrane surface. Hybridization (in a solution Containing Nucleic acid probe) (Alkali treatment) antibody or enzyme (modified nucleotide labeled X-ray film(radio- actively labeled ) Wash to remove unhybri- dization probe and visualize Line up the hybridizated region or repeated hybridization
 (Alkali treatment) antibody or enzyme. (modified nucleotide labeled. X-ray film(radio- actively labeled ) Wash to remove unhybri- dization probe and visualize. Line up the hybridizated region or. repeated hybridization.")
76
探针的来源 1. 某种生物实验体系中分离到的基因序列,可作为从其它生物中分离相关基因的核酸探针。 2.根据蛋白质家族保守性,合成核酸探针。
功能相同或相近的蛋白质,存在着具有共同氨基酸序列的区段(保守区), 往往由彼此相邻的6~7个氨基酸组成。
, 往往由彼此相邻的6~7个氨基酸组成。")
77
寡核苷酸探针的人工合成 1. 探针的长度; 要使探针与目的基因序列严格互补,最少的探针长度15~16个核苷酸,实验中为保证足够的特异性,通常使用17~20个核苷酸。 2. 简并性; 根据蛋白质氨基酸组分合成的寡核苷酸探针,通常是混合物的形式。在这种探针库中,只有一种是与目的基因完全互补的。

78
由于探针库中只有一种是与目的基因完全互补的,其它的探针有可能与不相关的片断杂交,产生假阳性信号。 1.猜测体探针 2.PCR-猜测体探针

79
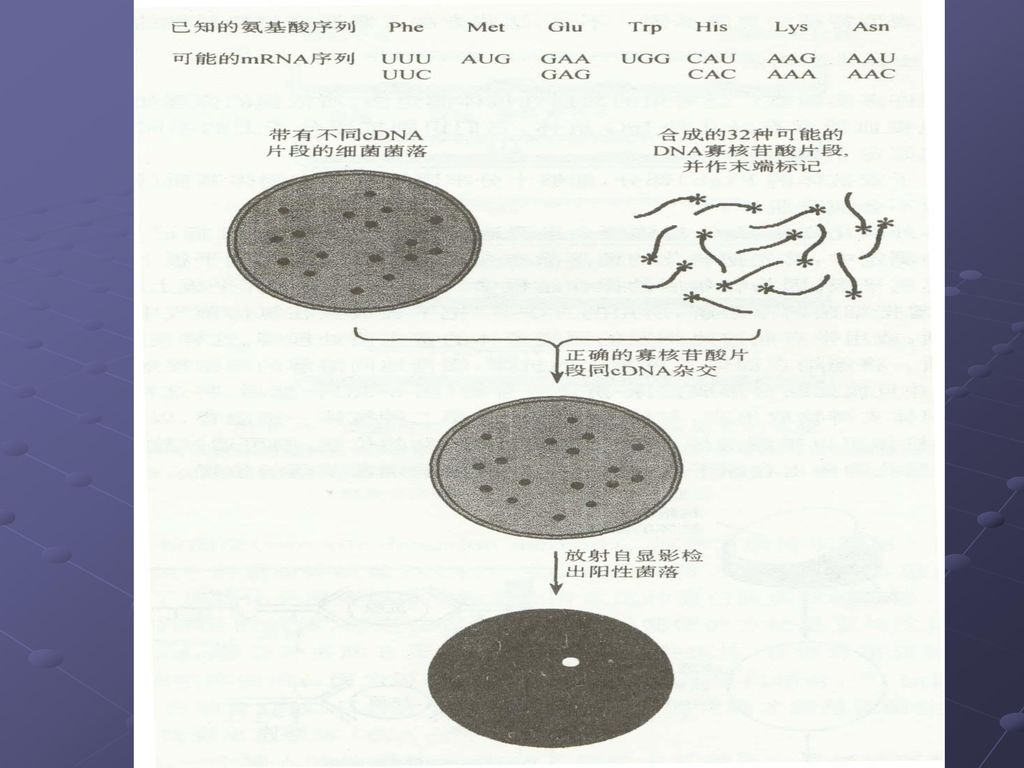
猜测体:一种人工合成的用于分离克隆基因的低简并性的寡核苷酸探针,其核苷酸序列是根据特定物种当中某种已知蛋白质的密码子使用频率,同时又采纳了根据猜测最可能在目的基因中出现的密码子资料,选择含有密码子简并程度最低的蛋白质区段进行合成。 一种较长的唯一的寡核苷酸序列,它能够大范围的却不能够完全的同靶序列互补。

81
根据末端32个氨基酸序列,设计 PCR简并引物,以cDNA为模板 用猜测探针杂交筛选阳性克隆 PCR-猜测体探针 (尿酸氧化酶基因)
")
82
应用差别杂交或扣除杂交法 分离克隆的目的基因

83
差别杂交(differential hybridization) 又称差别筛选 (differential screening ) 适用于分离经特殊处理而被诱发表达的mRNA的cDNA。 扣除杂交(subtractive hybridization ) 又叫扣除cDNA克隆( subtractive cDNA cloning) 通过构建扣除文库得以实现。
 又称差别筛选 (differential screening ) 适用于分离经特殊处理而被诱发表达的mRNA的cDNA。 扣除杂交(subtractive hybridization ) 又叫扣除cDNA克隆( subtractive cDNA cloning) 通过构建扣除文库得以实现。")
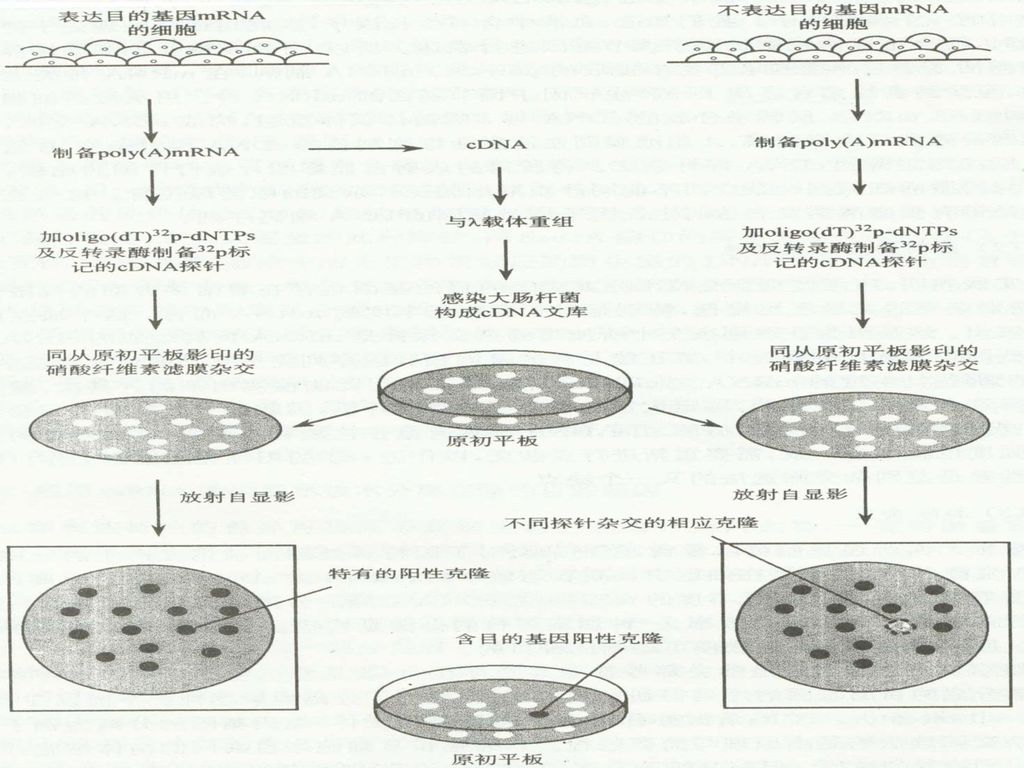
84
差别杂交: 需要两种不同的细胞群体(目的基因正常表达、目的基因不表达),分别制备两种不同mRNA提取物,以两种总mRNA探针平行杂交,对有表达目的基因的细胞总mRNA构建的克隆文库进行筛选。
,分别制备两种不同mRNA提取物,以两种总mRNA探针平行杂交,对有表达目的基因的细胞总mRNA构建的克隆文库进行筛选。")
86
差别杂交的局限性: 1. 杂交灵敏度低,对于低峰度的mRNA尤为明显 2
差别杂交的局限性: 1.杂交灵敏度低,对于低峰度的mRNA尤为明显 2.工作量大,重复性差,两套平行滤膜之间的DNA保有量有差别,导致杂交信号不一致。

87
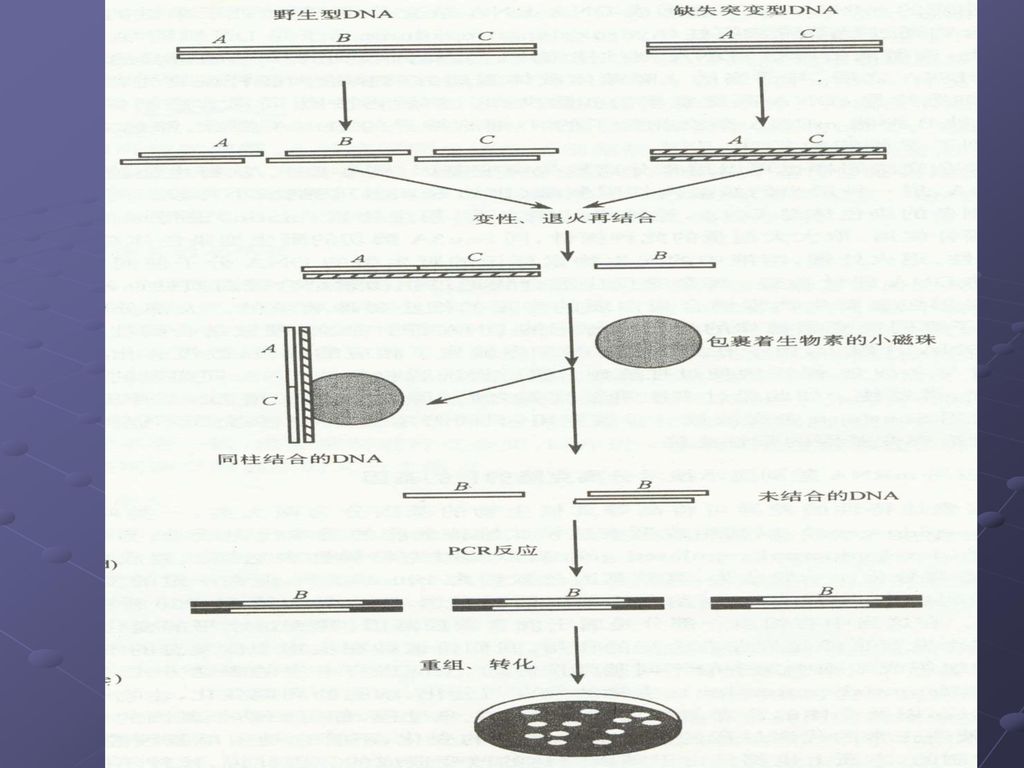
扣除杂交: 去除普遍共同存在的、或是非诱发产生的cDNA序列,从而使欲分离的目的基因的序列得到有效的富集。

89
应用mRNA差别显示技术(DDRT-PCR) 分离克隆的目的基因 (differential display reverse transcription polymerase chain reaction)
 分离克隆的目的基因 (differential display reverse transcription polymerase chain reaction)")
90
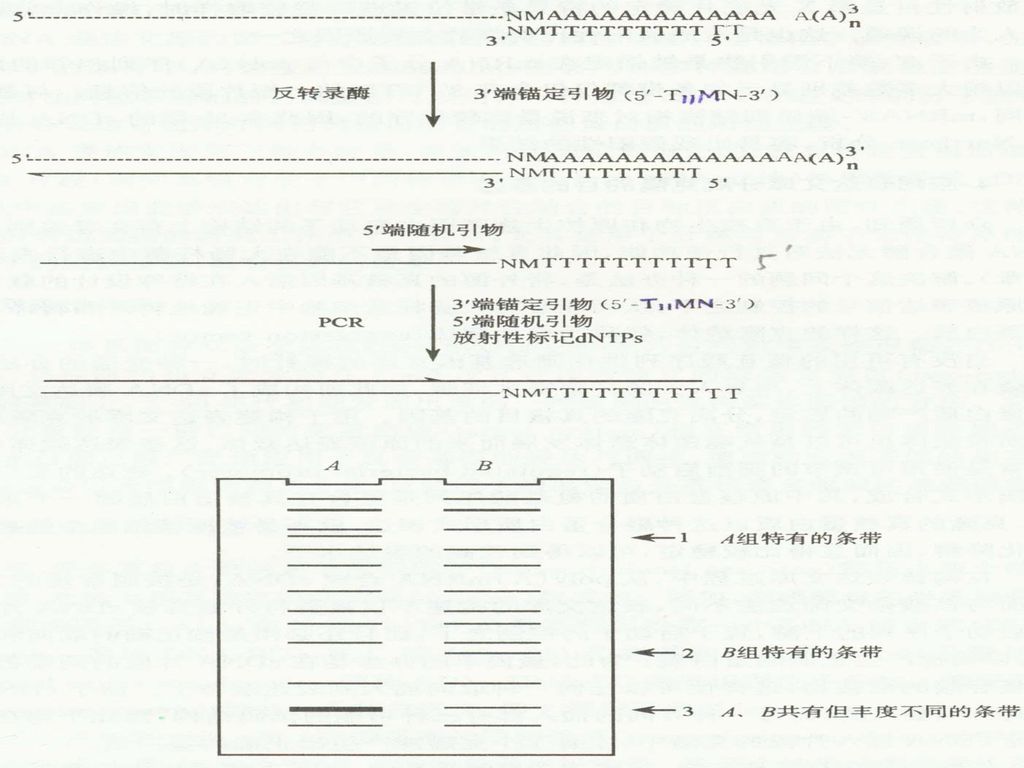
原理: 用3 ’-端锚定引物和反转录酶合成cDNA的第一链;用3 ’-端锚定引物和5’-端10-mer随机引物组成引物对,以反转录第一链为模板进行PCR扩增(DDRT-PCR),在标准的序列胶中电泳2~3小时,可显示出50~100条长度在100~5000bp之间的DNA条带。
,在标准的序列胶中电泳2~3小时,可显示出50~100条长度在100~5000bp之间的DNA条带。")
91
3 ’-锚定引物: P.Liang等人设计合成了全部12种不同的引物,用以反转录mRNA,合成第一链cDNA。这种引物通常叫作3'-端锚定脱氧核苷酸引物,并用5'-T11MN或5'-T12MN通式表示。其中M为除了T以外的任何一种核苷酸(即A、G或C),而N则为任何一种核苷酸(即A、G、C或T),故MN共有12种不同的排列组合方式。 mRNA: 5’-NMAAAAAA…AA-3’(12种序列) 3’-NMTTTTTTT…TT-5’
,而N则为任何一种核苷酸(即A、G、C或T),故MN共有12种不同的排列组合方式。 mRNA: 5’-NMAAAAAA…AA-3’(12种序列) 3’-NMTTTTTTT…TT-5’")
93
5’-随机引物: 10-mer,更好的同第一链结合,从而呈现出更多种的mRNA。一般用20种随机引物和12种3’-端锚定引物组成的全部240组引物对PCR扩增后,所产生的大约20 000条条带,基本涵盖了在一定的发育阶段某种类型细胞中所表达的全部的mRNA。

94
DDRT-PCR(mRNA differential disply reverse transcription polymerase chain reaction) 由于在cDNA群体中,代表特定细胞类型或发育阶段的mRNA的含量非常低,所以要把一种只在某一阶段或某种细胞类型中表达、而在另一发育阶段或某种细胞类型中不表达的目的基因分离出来,就需要PCR扩增。
 由于在cDNA群体中,代表特定细胞类型或发育阶段的mRNA的含量非常低,所以要把一种只在某一阶段或某种细胞类型中表达、而在另一发育阶段或某种细胞类型中不表达的目的基因分离出来,就需要PCR扩增。")
95
基本过程: 1. 从一对处于不同发育阶段(或不同基因型) 的细胞群体中分离总mRNA,并用3’-端锚定引物作反转录合成第一链cDNA; 2
基本过程: 1. 从一对处于不同发育阶段(或不同基因型) 的细胞群体中分离总mRNA,并用3’-端锚定引物作反转录合成第一链cDNA; 2.用5’-端随机引物和3’-端锚定引物组成的引物对,在加入放射性同位素标记的dNTP的条件下,以第一步反转录产物作模板进行PCR扩增。 3.将扩增样品在变性的DNA测序胶中进行电泳分离;
 的细胞群体中分离总mRNA,并用3’-端锚定引物作反转录合成第一链cDNA; 2.用5’-端随机引物和3’-端锚定引物组成的引物对,在加入放射性同位素标记的dNTP的条件下,以第一步反转录产物作模板进行PCR扩增。 3.将扩增样品在变性的DNA测序胶中进行电泳分离;")
96
4. 将有关的差别表达的DNA条带从测序胶上切割下来回收其DNA片断; 5. 胶块中的DNA量非常少,不能用于克隆,需进行二次扩增; 6
4.将有关的差别表达的DNA条带从测序胶上切割下来回收其DNA片断; 5.胶块中的DNA量非常少,不能用于克隆,需进行二次扩增; 6.将克隆的特定的DNA分别同基因组DNA及总mRNA作Southern或Northern杂交,测序; 7.以此目的片断作探针从cDNA文库中或基因组文库中筛选全长的cDNA克隆或基因组克隆。

98
优点: 1.可以同时比较多个样品表达的差异; 2.可以同时检测“上游”及“下游”的基因;
3.检测灵敏度高,所需样品少,经PCR扩增一些低峰度的mRNA也可以被检测出来; 4.结合使用了PCR和序列胶电泳分析两项技术,使本方法显得较单方便。

99
局限性: 1.假阳性比例高(50%~75%); 同一长度的条带,含有多种DNA序列;邻近条带回收时,造成人为误差;
mRNA3’-端序列保守性高,而常用3’-端cDNA作探针。 2.扩增的差别条带分子长度比较短小(110~450bp);
;")
100
应用表达文库 分离克隆的目的基因

101
表达文库分离克隆的目的基因 当没有可用的核苷酸序列供作筛选基因文库的探针时,将cDNA克隆在表达载体上,再导入大肠杆菌寄主细胞然后通过对蛋白质产物的鉴定,分离克隆的真核目的基因。

102
特点: 1.表达的蛋白质以融合蛋白质的形式存在,其中原核蛋白质的氨基酸序列是整合在真核蛋白质的一个末端,不易被原核细胞中的有关蛋白酶消化降解,因而显得比较稳定,可以得到较高的表达水平。 2. cDNA片断必须置于启动子序列下游,在其控制之下;按正确的取向和读码结构插入,确保产生正确的蛋白质。

103
筛选的方法: 1. 利用抗体筛选表达文库; 2. 测定蛋白质的功能;
3. 用放射性同位素标记的、带有特定蛋白质结合位点的DNA片断作探针筛选表达文库。

104
将菌落或噬菌斑原位复制到膜上 处理之后蛋白质暴露,与第一抗体温育 漂洗未结合的抗体,加入经标记的第二抗体 能与第一抗体结合 抗体筛选表达文库

105
生物化学研究表明,存在Ca++离子的条件下,钙调蛋白能与许多种酶结合形成稳定的复合物。
2.测定蛋白质的功能 生物化学研究表明,存在Ca++离子的条件下,钙调蛋白能与许多种酶结合形成稳定的复合物。 将放射性同位素标记得钙调蛋白用作探针筛选cDNA表达文库,鉴定能够表达出与它特异性结合的目标蛋白质的阳性克隆。

106
3.用放射性同位素标记的、带有特定蛋白质结合位点的DNA片断作探针筛选表达文库 鉴定能够表达出与特定DNA片断(真核启动子中与转录因子结合的DNA元件)结合的目标蛋白质的克隆。
结合的目标蛋白质的克隆。")
107
酵母双杂交体系 (two-hybrid system)
")
108
酵母双杂交体系又叫相互作用陷阱 (interaction trap) 有效的分离能与已知的靶蛋白质相互作用的蛋白质的编码基因。
应用于真核基因地表达调控,细胞粘合因子间的相互作用、信号转导通路以及细胞周期与分化、反式因子的鉴定与分离等研究

109
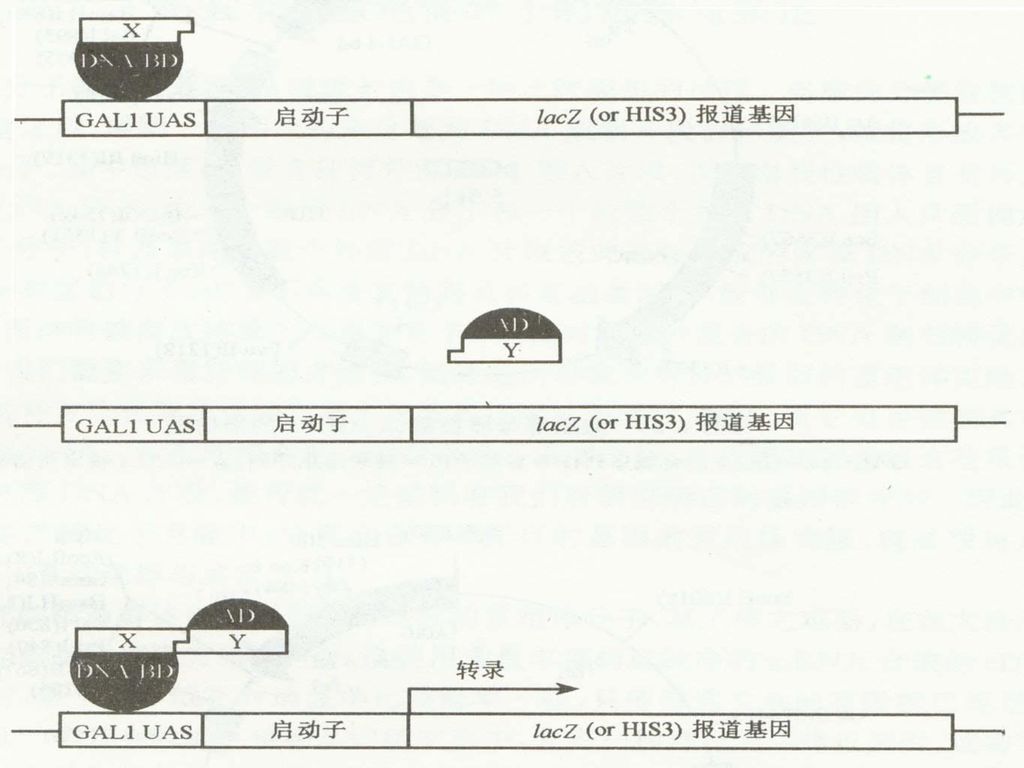
基本原理: 许多真核生物的转录激活因子都是由两个结构上可以分开的、功能上相互独立的结构域组成(example); 应用重组DNA技术,也可以将来自同一个转录因子的、或者两种不同转录因子的、分开的两种结构域,在体内重新组装成具有功能的转录因子,从而激活UAS(上游激活序列)下游启动子调节的报告基因的表达。
; 应用重组DNA技术,也可以将来自同一个转录因子的、或者两种不同转录因子的、分开的两种结构域,在体内重新组装成具有功能的转录因子,从而激活UAS(上游激活序列)下游启动子调节的报告基因的表达。")
110
Example: 酿酒酵母的半乳糖苷酶基因的转录激活因子GAL4,在N端1~147位氨基酸区段有一个结合域( DNA-binding domain, DNA-BD ); 在C端768~881位氨基酸区段有一个转录激活域(transcription activation domain,AD)DNA -BD 能识别激活序列并与之结合;AD通过同转录机制中的其它成份之间的结合作用启动下游基因进行转录。 用DNA重组技术将它们分开,即使在同一个寄主中表达,也不会直接发生相互作用不能激活相关的效应基因进行转录。
; 在C端768~881位氨基酸区段有一个转录激活域(transcription activation domain,AD)DNA -BD 能识别激活序列并与之结合;AD通过同转录机制中的其它成份之间的结合作用启动下游基因进行转录。 用DNA重组技术将它们分开,即使在同一个寄主中表达,也不会直接发生相互作用不能激活相关的效应基因进行转录。")
111
寄主菌株:剔除掉GAL4基因的酿酒酵母 1. SFY526和HF7c带有报告基因lacZ、 HIS3和LEU2
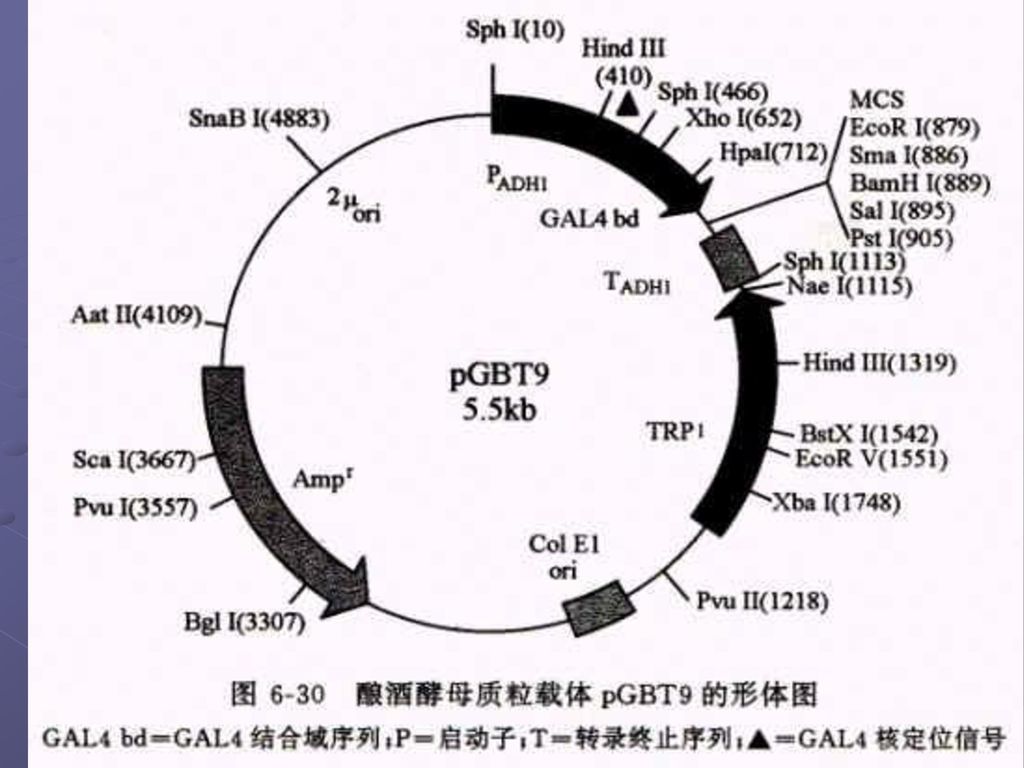
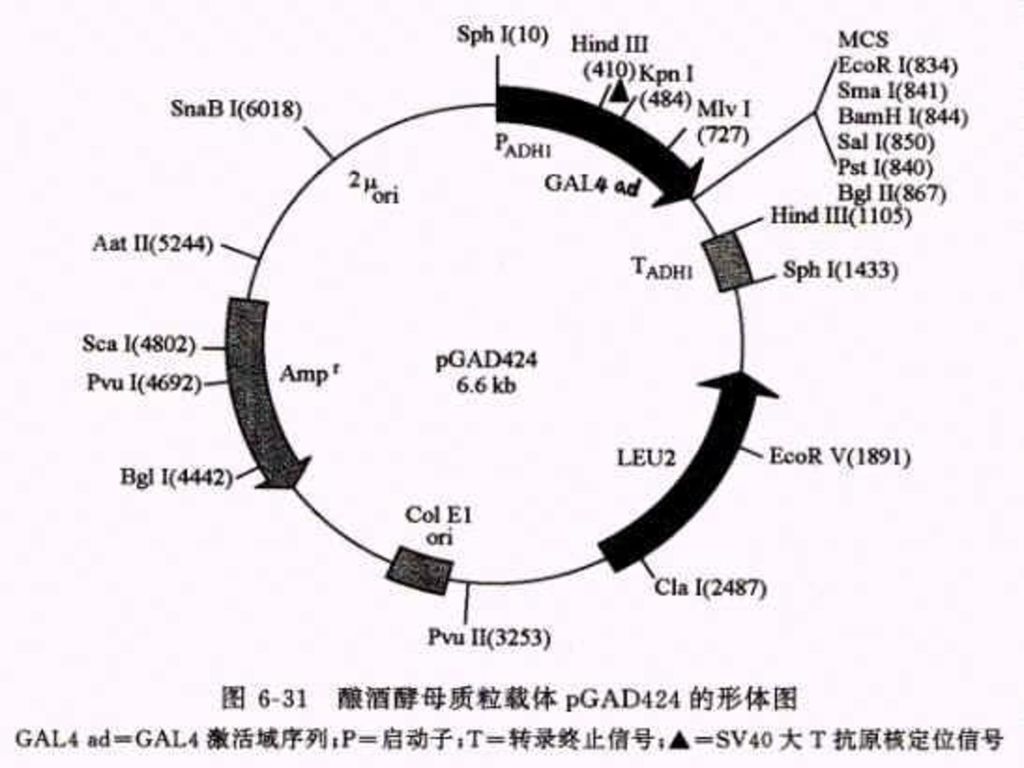
2.还有两种可以在大肠杆菌和酵母中自主复制的穿梭载体 a. pGBT9(DNA-BD):靶基因是按正确的取向和读码结构被克隆在载体的多克隆位点区内,于是靶蛋白和GAL4-BD之间产生融合作用,形成杂种蛋白质 b.第二种质粒载体pGAD424(AD质粒载体)用来构建cDNA表达文库专用载体,克隆cDNA片断是按正确的取向和读码结构插入在 载体的多克隆位点区内,因此由cDNA 编码的蛋白质便会同GSL4-AD之间产生融合作用,形成杂种蛋白质。
:靶基因是按正确的取向和读码结构被克隆在载体的多克隆位点区内,于是靶蛋白和GAL4-BD之间产生融合作用,形成杂种蛋白质 b.第二种质粒载体pGAD424(AD质粒载体)用来构建cDNA表达文库专用载体,克隆cDNA片断是按正确的取向和读码结构插入在 载体的多克隆位点区内,因此由cDNA 编码的蛋白质便会同GSL4-AD之间产生融合作用,形成杂种蛋白质。")
115
酵母双杂交体系的主要实验过程: a. 选择缺失GAL4编码基因的酵母寄主菌株-SFY526或HF7c; b
酵母双杂交体系的主要实验过程: a. 选择缺失GAL4编码基因的酵母寄主菌株-SFY526或HF7c; b. 构建带有GAL1 UAS-启动子-lac Z(His3)的转化载体; c. 把已知的靶蛋白质编码基因克隆到pGBT9的多克隆位点上,把所有cDNA都克隆到pGAD424载体上,构成cDNA表达文库。 d. 从大肠杆菌中分别提取这两种重组质粒DNA,共转化感受态酿酒酵母菌株。 e. 将共转化的酵母菌株涂布于缺少Leu,Trp和His的培养基上,筛选表达相互作用的杂种蛋白的阳性菌落。
的转化载体; c. 把已知的靶蛋白质编码基因克隆到pGBT9的多克隆位点上,把所有cDNA都克隆到pGAD424载体上,构成cDNA表达文库。 d. 从大肠杆菌中分别提取这两种重组质粒DNA,共转化感受态酿酒酵母菌株。 e. 将共转化的酵母菌株涂布于缺少Leu,Trp和His的培养基上,筛选表达相互作用的杂种蛋白的阳性菌落。")
116
重组子分子的选择与鉴定 1. 遗传检测法 2. 物理检测法 3. 菌落或噬菌斑杂交筛选法 4. 免疫化学检测法 5. 蛋白质筛选法
6. 转译筛选法

117
遗传检测法 该方法要求克隆基因能够表达,并利用相应的基因突变型作为受体细胞,当受体细胞由突变型变为野生型时或赋予受体细胞特定的表型时,我们基本可以确定所克隆的基因是否是目的基因。 优点:简便、快速,结果较可靠。 缺点:基因必须表达并具有合适的受体 细胞。

118
1. 根据载体表型特征选择重组体分子的直接选择法 a. 抗药性记号插入失活选择法 b. β-半乳糖苷酶显色反应选择法

121
2. 根据插入序列的表型特征选择重组体分子的直接选择法
转化进来的外源DNA编码的基因,能够对大肠杆菌寄主菌株所具有的突变体发生体内抑制或互补效应,从而使被转化的寄主细胞表现出外源基因编码的表型特征。

122
Example: λgt. λB噬菌体(由于C片断的缺失而造成重组缺陷的λ red-噬菌体),在大肠杆菌lig ts菌株上生长不能形成噬菌斑,在有连接酶功能的大肠杆菌lig+菌株上形成噬菌斑。 将具有连接酶基因的重组体噬菌体λgt. λB,涂布在大肠杆菌lig ts菌株上时,通过寄主细胞的互补作用,能够形成噬菌斑。根据形成噬菌斑这种表型特征,直接选择具野生功能的重组体噬菌体。
,在大肠杆菌lig ts菌株上生长不能形成噬菌斑,在有连接酶功能的大肠杆菌lig+菌株上形成噬菌斑。 将具有连接酶基因的重组体噬菌体λgt. λB,涂布在大肠杆菌lig ts菌株上时,通过寄主细胞的互补作用,能够形成噬菌斑。根据形成噬菌斑这种表型特征,直接选择具野生功能的重组体噬菌体。 .")
123
缺点: 不单要求克隆的DNA片断必须大到括足以包含一个完整的基因,而且还要求所编码的基因能够在大肠杆菌中实现功能表达。 (对于真核基因很难达到要求)
")
124
物理检测法 1.凝胶电泳检测法 利用凝胶电泳的方法,鉴定外源片断是否插入及其长度。 优点:简便、快速。 缺点:只能提供克隆片段大小的信息。

125
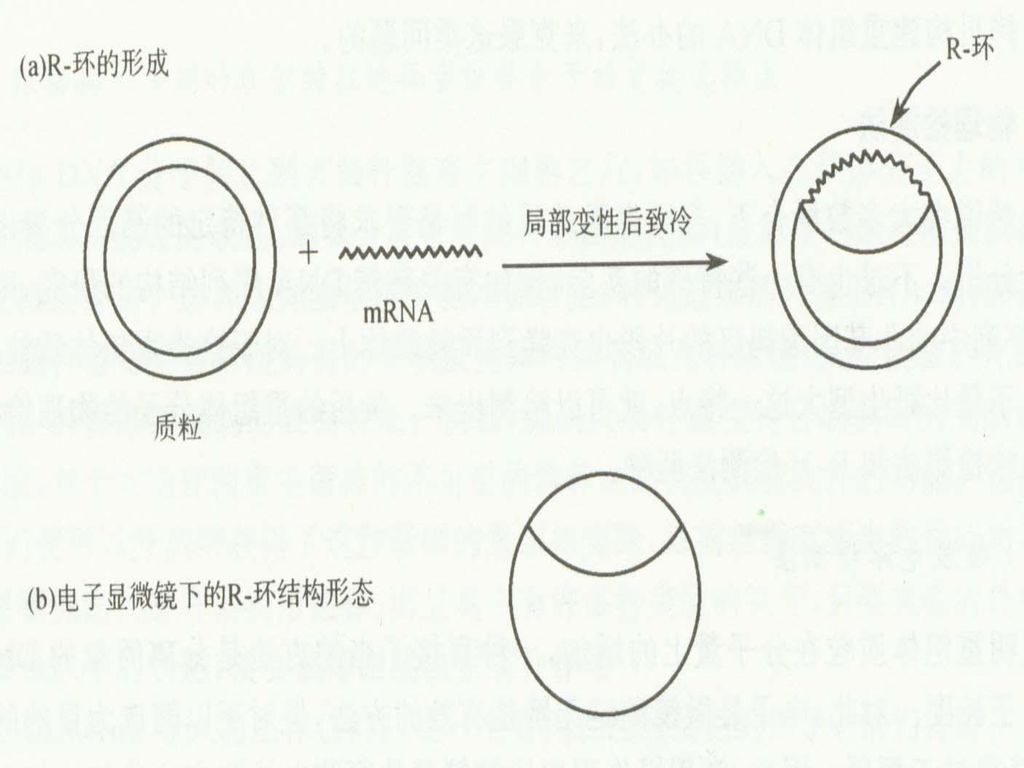
2. R-环检测法: 鉴定出双链DNA中存在的与特定RNA分子同源区域 在临近双链DNA变性温度下和高浓度的甲酰胺溶液中(70%),双链的DNA-RNA分子比双链的DNA-DNA分子更加稳定。 在这种条件下,RNA会同它的双链DNA分子中的互补序列退火形成稳定的RNA-DNA,而被取代的另一条DNA呈单链状态。这种由双链RNA-DNA和单链DNA形成的泡状体称R-环结构。
,双链的DNA-RNA分子比双链的DNA-DNA分子更加稳定。 在这种条件下,RNA会同它的双链DNA分子中的互补序列退火形成稳定的RNA-DNA,而被取代的另一条DNA呈单链状态。这种由双链RNA-DNA和单链DNA形成的泡状体称R-环结构。 .")
127
分子杂交技术检测法 包括菌落或噬菌斑的原位杂交技术。 优点:方法简便、快速。 缺点:必须具有相应的探针。

129
免疫化学检测法 利用免疫学的原理,检测克隆基因的表达产物。

130
1.放射性抗体检测法 该方法类似于原位杂交法。 优点:方法简便,一次可检测大量的 克隆子。 缺点:克隆基因必须表达并具有相应的 抗体。

131
原理: 1. 免疫血清含有几种IgG抗体,识别抗原分子上不同定子,并分别同各自识别的抗原定子相结合; 2
原理: 1. 免疫血清含有几种IgG抗体,识别抗原分子上不同定子,并分别同各自识别的抗原定子相结合; 2. 抗体分子或抗体的F(ab)部分,能够牢固的吸附在固体基质上(聚乙烯等塑料制品),而不容易被洗脱掉; 3. 通过体外碘化作用, IgG抗体会迅速的被放射性同位素125I标记上。
部分,能够牢固的吸附在固体基质上(聚乙烯等塑料制品),而不容易被洗脱掉; 3. 通过体外碘化作用, IgG抗体会迅速的被放射性同位素125I标记上。")
132
步骤: 1. 菌落溶解释放出抗原蛋白质; a. 把平板放置在氯仿蒸气中; b. 用烈性噬菌体的气溶胶喷洒; c

133
细菌菌落溶解,释放出抗原蛋白质

134
双位点检测法: 适用于含有杂种多肽菌落分析。 杂种多肽 A – B (A) 抗体 (B)抗体 固定在固体基质 125I标记
 抗体 (B)抗体 固定在固体基质 125I标记")
135
聚乙烯薄膜 滤膜 培养皿及菌落 放射性标记抗体检测法的操作程序 放射性标记抗体检测法的操作程序 抗体溶液

136
2.免疫沉淀检测法 在生长菌落的琼脂培养基中,加入专门抗这种蛋白质分子的特异性抗体。如果菌落的细菌会分泌出某种蛋白质,那么它的周围,就会出现一条由一种叫做沉淀素的抗体-抗原沉淀物所形成的白色的圆圈。 灵敏度低,易受干扰,实用性差。

137
3.表达载体产物之免疫化学检测法 用专门设计的表达载体,将真核基因编码的蛋白质在大肠杆菌中实现表达,通过免疫化学法进行检测。

139
蛋白质筛选法 用来检测同DNA特异性结合的蛋白质因子的一种方法。 操作程序:
1. 用硝酸纤维素滤膜进行“噬菌斑转移”,使其中的蛋白质吸附在滤膜上; 2. 将滤膜同放射性标记的含有DNA结合蛋白质编码序列的双链DNA寡核苷酸探针杂交; 3. 根据放射自显影结果筛选出阳性反应克隆。

140
转译筛选法 1. 杂交抑制的转译 该方法用于cDNA文库中目的基因的筛选。其原理是在体外无细胞的转译中利用cDNA与mRNA杂交后导致mRNA的不能翻译,从而筛选阳性克隆子。 分离的基因编码着丰富的mRNA。 优点:不需要克隆基因的表达,不需要探针或抗体。 缺点:较烦琐。

141
步骤: 1. 将从大肠杆菌菌落或噬菌体群体中制备的带有目的基因 的重组质粒DNA变性后,与总mRNA杂交; 2
步骤: 1. 将从大肠杆菌菌落或噬菌体群体中制备的带有目的基因 的重组质粒DNA变性后,与总mRNA杂交; 2. 转入无细胞转译体系(麦胚提取物或兔网织红细胞提取物),由于加有35S标记的蛋氨酸,转译的多肽蛋白质可以通过聚丙烯酰胺凝胶电泳和放射自显影进行分析; 3. 结果同未杂交的mRNA转译产物作比较,从中找出其转译合成被抑制的mRNA,即同目的基因变性DNA互补彼此杂交的mRNA。
,由于加有35S标记的蛋氨酸,转译的多肽蛋白质可以通过聚丙烯酰胺凝胶电泳和放射自显影进行分析; 3. 结果同未杂交的mRNA转译产物作比较,从中找出其转译合成被抑制的mRNA,即同目的基因变性DNA互补彼此杂交的mRNA。")
142
2. 杂交选择的转译 是一种直接的选择法,比杂交抑制的转译要敏感得多,而且还可适用于低峰度的mRNA之cDNA重组分子的检测。

143
步骤: 1. 将重组体库中分离的克隆DNA,固定在消化纤维素膜上,同总mRNA杂交; 2. 分离杂交的mRNA; 3

Similar presentations




:核蛋白体组成成分 转移 RNA ( tRNA ):转运氨基酸 信使 RNA ( mRNA ):蛋白质合成模板 不均一核 RNA ( hnRNA ):成熟 mRNA 的前体 小核 RNA ( snRNA ):>")



>")







 从20世纪中叶开始,分子生物学研究得到高速发展,除了基础理论的重大突破,主要原因之一是研究方法,特别是基因操作和基因工程技术的进步。>")




 A.细菌的遗传物质主要是DNA B.病毒的遗传物质主要是RNA>")

