Download presentation
Presentation is loading. Please wait.

1
基因组数据注释和功能分析 陈启昀 陈 辰 丁文超 张增明 浙江加州国际纳米技术研究院(ZCNI)
")
2
课程内容 基因组学 系统生物学 转录物组学 蛋白质组学 实习一 基因组数据注释和功能分析 实习二 核苷酸序列分析 实习三
芯片的基本数据处理和分析 实习四 蛋白质结构与功能分析 实习五 蛋白质组学数据分析 实习六 系统生物学软件实习 基因组学 系统生物学 转录物组学 蛋白质组学

3
课程提纲 通过序列比对工具BLAST学习,了解蛋白编码基因的功能注释原理 介绍多序列联配工具ClustalX
分子进化分析软件MEGA4的基本知识,掌握系统发生树绘制的基本方法

4
序列比对的进化基础 什么是序列比对: 将两个或多个序列按照最佳匹配方式排列在一起。 对应的相同或相似的符号排列在同一列上。
错配与突变相应,空位与插入或缺失对应。 序列比对的目的: 从核酸以及氨基酸的层次去分析序列的相同点和不同点,以推测他们的结构、功能以及进化上的联系 通过判断两个序列之间的相似性来判定两者是否具有同源性 相似性:可以被数量化,如:序列之间相似部分的百分比 同源性:质的判断,两个基因在进化上是否曾有共同祖先的推断

5
BLAST 基本局部比对搜索工具(Basic Local Alignment Search Tool) NCBI上BLAST服务的网址:
NCBI上BLAST程序的下载: ftp://ftp.ncbi.nlm.nih.gov/blast/executables/release/ NCBI的BLAST数据库下载网址: ftp://ftp.ncbi.nlm.nih.gov/blast/db/

6
选择物种 选择blast程序

7
QuerySequence AminoacidSequence DNASequence BLASTp tBLASTn BLASTn
BLASTx tBLASTx Translated Translated Translated Protein Database Nucleotide Database Nucleotide Database Protein Database Nucleotide Database

8
程序名 搜索序列 数据库 内容 备注 blastp Protein 比较氨基酸序列与蛋白质数据库 使用取代矩阵寻找较远的关系,进行SEG过滤
blastn Nucleotide 比较核酸序列与核酸数据库 寻找较高分值的匹配,对较远的关系不太适用 blastx 比较核酸序列理论上的六个读码框的所有转换结果和蛋白质数据库 用于新的DNA序列和ESTs的分析,可转译搜索序列 tblastn 比较蛋白质序列和核酸序列数据库,动态转换为六个读码框的结果 用于寻找数据库中没有标注的编码区,可转译数据库序列 tblastx 比较核酸序列和核酸序列数据库,经过两次动态转换为六个读码框的结果 转译搜索序列与数据库序列

9
以Blastx为例: 目标序列为ATG AGT ACC GCT AAA TTA GTT AAA TCA AAA GCG ACC AAT CTG CTT TAT ACC CGC 6个读码框翻译 5’端到3’端 第一位起始: ATG AGT ACC GCT AAA TTA GTT AAA TCA AAA GCG ACC AAT CTG CTT TAT ACC CGC 第二位起始: TGA GTA CCG CTA AAT TAG TTA AAT CAA AAG CGA CCA ATC TGC TTT ATA CCC GC 第三位起始: GAG TAC CGC TAA ATT AGT TAA ATC AAA AGC GAC CAA TCT GCT TTA TAC CCG C 3’端到5’端 GCG GGT ATA AAG CAG ATT GGT CGC TTT TGA TTT AAC TAA TTT AGC GGT ACT CAT CGG GTA TAA AGC AGA TTG GTC GCT TTT GAT TTA ACT AAT TTA GCG GTA CTC AT GGG TAT AAA GCA GAT TGG TCG CTT TTG ATT TAA CTA ATT TAG CGG TAC TCA T

11
与核酸相关的数据库 与蛋白质相关的数据库

12
BlastN 序列或目标序列的GI号 以文件格式上传 选择数据库

13
配对与错配 空位罚分

14
BlastP PSI-BLAST 可以幫使用者比對相同蛋白質家族的氨基酸序列 PHI-BLAST 可以就特定的氨基酸部分進行比對;

15
打分矩阵: PAM30 PAM70 BLOSUM80 BLOSUM62 BLOSUM45

16
选择打分矩阵(scoring matrix)
The PAM family Based on global alignments The PAM1 is the matrix calculated from comparisons of sequences with no more than 1% divergence. Other PAM matrices are extrapolated from PAM1. The BLOSUM family Based on local alignments. BLOSUM62 is a matrix calculated from comparison s of sequences with no less than 62% divergence. All BLOSUM matrices are based on observed alignments ;they are not extrapolated from comparisons of closely related proteins. Pam 1 表示有1%的氨基酸形成突变 Blosum62 表示比对结果中至少有62%的氨基酸相同

17
进行比对的数据库 图形化结果

18
E值(E-value)表示仅仅因为随机性造成获得这一 比对结果的可能性。这一数值越接近零,发生这一事件的可能性越小。
表示仅仅因为随机性造成获得这一 比对结果的可能性。这一数值越接近零,发生这一事件的可能性越小。")
20
上机实习1:网上运行blastx和blastn
(NCBIblast网址: >lesson.seq.screen.Contig34 TTTTTTTTTTTTTTTTTAGTGCCAGTTTTTTTTTTTATTTGTAAAGCTCTGCCATAAACTTCTAGCGTGTGCCAATGGTCACCTGCCACACTCGCACCAGGTTGTCCGTGTAGCCAGCAAACAGAGTCTGGCCATCAGCAGACCAGGCCAGGGAGGTGCACTGGGGTGGTTCTGCCTTGCTGCTGGTACTGATAACTTCTTGCTTCAGTTCATCTACAATGATCTTTCCCTCTAAATCCCAGATCTTGATGCTGGGGCCTGTGGAGCACACAGCCAGTAGCGGTTAGGGCTGAAGCACAGGGCGTTGATGATGTCCCCACCATCTAGCGTGTAAAGGTGTTTGCCTTCGTTGAGATCCCATAACATGGCCTGGCCATCCTTGCCTCCAGAAGCACAGAGGGATCCATCTGGAGAGACAGTCACCGTGTTCAGATAGCCTGTGTGGCCAATGTGGTTGGTCTTCAGCTTGCAGTTAGCCAGGTTCCATACCTTGACCAGCTTGTCCCAGCCACAGGAGACGATGATAGGGTTGCTGCTGTTGGGCGAGAAGCGGACACAAGACACCCACTCTGAGTGGCTCTCATCCTGGACAGTGTATTTGCACACACCCAGGGTATTCCATAGCTTGATGGTTTTATCTCGAGATCCAGAGACAATCTGCCGGTTGTCAGAGGAGAAGGCCACACTCAGCACATCCTTGGTATGGCCCACAAATCGCCTCGTGGTGGTGCCCGTTGTGAGATCCCAGAAGGCGCAGGGTTCCATCCCAGGAGCCTGAGAGGGCAAACTGGCCATCTGAGGAGATAACCACATCACTAACAAAGTGGGAGTGACCCCGCAGAGCACGCTGTGGAATTCCATAGTTGGTCTCATCCCTGGTCAGTTTCCACATGATGATGGTCTTATCTCGAGAGGCGGAGAGGATCATGTCCGGGAACTGCGGGGTAGTAGCGATCTGGGTTACCCAGCCGTTGTGGCCCTTGAGGGTGCCACGAAGGGTCATCTGCTCAGTCATGGCGGCGGCGAGAGCGTGTTCGCTGCAGCGACGAGGATGGCACTGGATGGCTTAGAGAAACTAGCACCACAGTCGACC 对contig34进行网上blastn(演示), blastx(自行操作)比对
, blastx(自行操作)比对.")
21
本地运行BLAST 下载NCBI上blast程序:
ftp://ftp.ncbi.nlm.nih.gov/blast/executables/release/ 安装(安装到C:\) 数据库的格式化(formatdb) 程序运行(blastall)
 数据库的格式化(formatdb) 程序运行(blastall)")
22
登陆NCBI的FTP下载blast程序

23
将数据库文件(db)及目标序列文件(in)保存在Blast/bin文件夹下
双击安装到C盘 产生三个文件夹 bin data doc bin含可执行程序(将数据库及需要比对操作的数据放入该文件); data文件夹含打分矩阵及演示例子的序列数据信息; doc文件夹含关于各子程序的说明文档。 将数据库文件(db)及目标序列文件(in)保存在Blast/bin文件夹下
; data文件夹含打分矩阵及演示例子的序列数据信息; doc文件夹含关于各子程序的说明文档。 将数据库文件(db)及目标序列文件(in)保存在Blast/bin文件夹下.")
24
本地数据库的构建 查看db文件 由fasta格式的序列组成

25
数据库的格式化 formatdb命令用于数据库的格式化: formatdb [option1] [option2] [option3]…
-i database_name 需要格式化的数据库名称 -p T\F 待格式化数据库的序列类型 (核苷酸选F;蛋白质选T;默认值为T) 例:formatdb -i db -p T 对蛋白质数据库“db”进行格式化
![数据库的格式化 formatdb命令用于数据库的格式化: formatdb [option1] [option2] [option3]…](http://slidesplayer.com/slide/14001822/86/images/25/%E6%95%B0%E6%8D%AE%E5%BA%93%E7%9A%84%E6%A0%BC%E5%BC%8F%E5%8C%96+formatdb%E5%91%BD%E4%BB%A4%E7%94%A8%E4%BA%8E%E6%95%B0%E6%8D%AE%E5%BA%93%E7%9A%84%E6%A0%BC%E5%BC%8F%E5%8C%96%EF%BC%9A+formatdb+%5Boption1%5D+%5Boption2%5D+%5Boption3%5D%E2%80%A6.jpg "-i database_name 需要格式化的数据库名称. -p T\F 待格式化数据库的序列类型. (核苷酸选F;蛋白质选T;默认值为T) 例:formatdb -i db -p T. 对蛋白质数据库 db 进行格式化.")
26
程序运行 blastall命令用于运行五个blast子程序: blastall [option1] [option2] [option3]
*可在dos下输入blastall查看各个参数的意义及使用 blastall常用参数 四个必需参数 -p program_name,程序名,根据数据库及搜索文件序列性质进行选择; -d database_name,数据库名称,比对完成格式化的数据库; -i input_file,搜索文件名称; -o output_file,BLAST结果文件名称; 两个常用参数 -e expectation,期待值,默认值为10.0,可采用科学计数法来表示,如2e-5; -m alignment view options:比对显示选项,其具体的说明可以用以下的比对实例说明 例:blastall -p blastx -d db -i in -o out -e 2e-5 -m 9 (表格显示比对结果) 采用blastx程序,将in中的序列到数据库bd中进行比对,结果以表格形式输入到out文件
![程序运行 blastall命令用于运行五个blast子程序: blastall [option1] [option2] [option3]](http://slidesplayer.com/slide/14001822/86/images/26/%E7%A8%8B%E5%BA%8F%E8%BF%90%E8%A1%8C+blastall%E5%91%BD%E4%BB%A4%E7%94%A8%E4%BA%8E%E8%BF%90%E8%A1%8C%E4%BA%94%E4%B8%AAblast%E5%AD%90%E7%A8%8B%E5%BA%8F%3A+blastall+%5Boption1%5D+%5Boption2%5D+%5Boption3%5D.jpg "*可在dos下输入blastall查看各个参数的意义及使用. blastall常用参数. 四个必需参数. -p program_name,程序名,根据数据库及搜索文件序列性质进行选择; -d database_name,数据库名称,比对完成格式化的数据库; -i input_file,搜索文件名称; -o output_file,BLAST结果文件名称; 两个常用参数. -e expectation,期待值,默认值为10.0,可采用科学计数法来表示,如2e-5; -m alignment view options:比对显示选项,其具体的说明可以用以下的比对实例说明. 例:blastall -p blastx -d db -i in -o out -e 2e-5 -m 9 (表格显示比对结果) 采用blastx程序,将in中的序列到数据库bd中进行比对,结果以表格形式输入到out文件.")
27
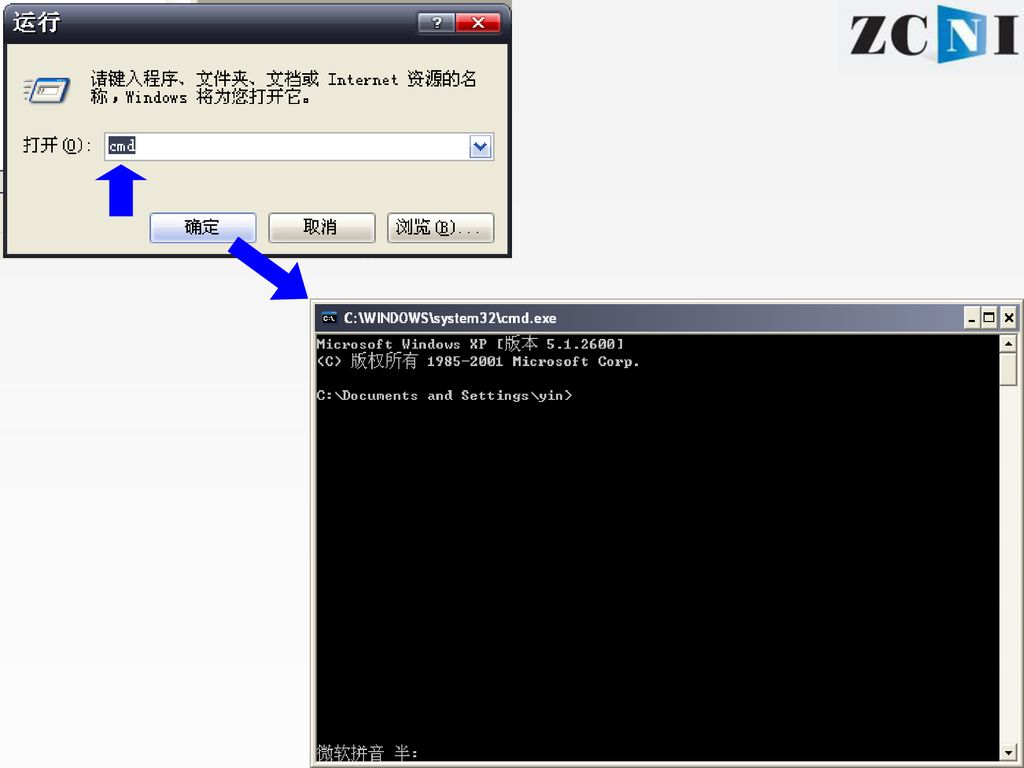
上机实习2:本地运行blastx 进入DOS命令行提示符状态(“运行”cmd) 进入C盘“cd\”
进入包含序列数据的bin目录下“cd blast\bin” 察看目录下内容“dir” 格式化数据库db“formatdb -i db -p T” 运行blastx “blastall -p blastx -i in -d db -o out -e 2e-5 -m 9 ” 察看结果“more out ”或在 windows下双击打开 输入 数据库类型:F/T Blast程序 序列输入 数据库 结果输出

29
输入“cd\”-〉回车 回到安装目录C盘 输入“cd blast\bin”-〉回车 到达blast程序下bin文件夹

30
输入“dir”-〉回车 察看bin文件夹下内容 bin文件夹下包含以.exe为后缀的程序文件以及这次实习需要用到的数据可文件“bd”和目标序列文件“in”

31
输入“more db”-〉回车察看db文件内容
空格键翻页 输入“q”跳出 输入“more db”-〉回车察看db文件内容

32
输入“formatdb -i db -p T”-〉回车

33
输入“dir”-〉回车 察看bin文件夹下内容 格式化以后产生的文件

34
输入“blastall -p blastx -i in -d db -o out -e 2e-5 -m 9”
-〉回车 运行blastx程序

35
产生的结果文件“out”

36
用”more out” 察看结果文件

37
不使用-m参数时 比对结果显示序列两两比对

38
用”more out” 察看结果文件

39
多序列比对的目的 从物种的一些分子特性出发,从而了解物种之间的生物系统发生的关系。
通过序列同源性的比较进而了解基因的进化以及生物系统发生的内在规律。

40
多序列比对的应用: 系统发育分析(phylogenetic analysis) 结构预测(structure prediction) 序列基序鉴定(sequence motif identification) 功能预测(function prediction) ClustalW/ClustalX:一种全局的多序列比对程序,可以用来绘制亲缘树,分析进化关系。 MEGA4
 ClustalW/ClustalX:一种全局的多序列比对程序,可以用来绘制亲缘树,分析进化关系。 MEGA4.")
41
ClustalW/X的运行 本地运行 命令行操作的Clustal W(linux & windows)
窗口化操作的ClustalX(windows) 下载页面:ftp://ftp.ebi.ac.uk/pub/software/ 欧洲生物学中心(EBI)还提供了Clustal W的网上运行服务(
 下载页面:ftp://ftp.ebi.ac.uk/pub/software/ 欧洲生物学中心(EBI)还提供了Clustal W的网上运行服务(")
42
下载ClustalX 各种参数设定 目标序列

43
Jalview 结果下载

44
点击Start Jalview打开java程序窗口

45
上机实习3:本地运行ClustalX 17-RNASE1.fasta 多序列比对 (Multiple Alignment)
")
46
在 C:\zcni\实习1\Clustalx2 文件夹下,找到clustalx.exe 双击打开

47
ClustalX窗口

48
点击File下拉菜单中 Load sequences选项, 打开序列文件17-RNASE1.fasta.txt

49
打开后的界面

50
点击进行多序列比对

51
可在Alignment下拉菜单中的Alignment Parameters中设定各个参数

52
点击Alignment下拉菜单中的Do Complete Alignment进行比对

53
比对结果 “*”、“:”、“.” 和空格依次代表改位点的序列一致性由高到低

54
MEGA4 一个关于序列分析及比较 统计的工具包 包含距离建树,MP等建 树法 自动或手动进行序列比对; 推断进化树;
估算分子进化率,进行进化假设测验; 联机进行数据库搜索; …

55
MEGA4可以识别fasta格式文件 将 17-RNASE1.fasta.txt 重命名为 17-RNASE1.fasta

56
选择打开方式为MEGA4,打开17-RNASE1.fasta,自动跳出序列窗口
用ClustalW做多序列联配

57
ClustalW参数设置

59
以.meg格式保存结果

60
回到MEGA主窗口 激活所保存的文件(.meg)
")
61
编辑标注 保守区域标注 不匹配的区域

62
回到MEGA4主窗口构建进化树 已被激活的文件

63
选择Bootstrap重复次数,至少为100次

64
四种系统进化树构建方法 分化程度较大的远缘序列: 邻位相连法(neighbor-joining,NJ) 最小进化法(ME)
分化程度较小的近缘序列: 最大简约法(MP) 除权配对法(UPGMA)
 除权配对法(UPGMA)")
65
进化树的可靠性分析 BootstrapMethod 从排列的多序列中随机有放回的抽取某一列,构成相同长度的新的排列序列 重复上面的过程,得到多组新的序列 对这些新的序列进行建树,再观察这些树与原始树是否有差异,以此评价建树的可靠性 至少进行100次重复取样

66
原始数据多 序列比对结果 对序列中每个 位置重复抽样, 基于原比对结果 生成多个样本

67
Original tree Bootstrap consensus tree 节点上的值为通过 Bootstrap检验的次数

68
不同树型 Tree:树型选择 Branch:分支信息修改 Label:分支名称修改 Scale:标尺设定 Cutoff:cutoff值

69
软件 网址 说明 ClustalX 图形化的多序列比对工具 ClustalW 命令行格式的多序列比对工具 GeneDoc 多序列比对结果的美化工具 BioEdit 序列分析的综合工具 MEGA 图形化、集成的进化分析工具,不包括ML PAUP 商业软件,集成的进化分析工具 PHYLIP 免费的、集成的进化分析工具 PHYML 最快的ML建树工具 PAML ML建树工具 Tree-puzzle 较快的ML建树工具 MrBayes 基于贝叶斯方法的建树工具 MAC5 TreeView 进化树显示工具

70
上机练习4:MEGA4.0

71
谢谢!

72
选择构树方法 最大简约法(maximumparsimony,MP)
对所有可能的拓扑结构进行计算,并计算出所需替代数最小的那个拓扑结构,作为最优树。 基于距离矩阵 UPGMA(UnweightedPair-GroupMethodusingAnathematicAverage) 将类间距离定义为两个类成员距离的平均值,广泛应用于距离矩阵 NJ(Neighbor-joining) 把所有n个序列两两比对,构建NJ树(起指导作用),每个对比后的成对序列都可以跟第三条序列或者另一个新的alignment比对,按照距离远近,用来决定下一个参与 比对的序列
 将类间距离定义为两个类成员距离的平均值,广泛应用于距离矩阵. NJ(Neighbor-joining) 把所有n个序列两两比对,构建NJ树(起指导作用),每个对比后的成对序列都可以跟第三条序列或者另一个新的alignment比对,按照距离远近,用来决定下一个参与 比对的序列.")
73
最大简约法(MP) 不需要处理大量核苷酸或者氨基酸替代 存在较多的回复突变或平行突变,而被检验的序列位点数又比较少的时候,可能会给出一个不合理的或者错误的进化树推导结果 UPGMA 所有分支突变率相近 突变率相差较大时(现已较少使用) 邻接法(NJ) 远源序列 对相似度很低的序列,往往出现Long-branch attraction(LBA,长枝吸引现象),严重干扰进化树的构建
 邻接法(NJ) 远源序列. 对相似度很低的序列,往往出现Long-branch attraction(LBA,长枝吸引现象),严重干扰进化树的构建.")
Similar presentations



有限责任公司和包头稀土研究院的建成与 发展,这里又被称作稀土之都。 包头稀土研究院 包 头位于内蒙古自治区中部,东与呼和浩特市相邻,西与巴彦 淖尔盟市连接 ,北与蒙古国接壤.>")







 ——把握人事,洞察百态 补上一课 如何读懂小说 第1讲 情节 第2讲 人物 第3讲 环境 >")





.>")




