运动神经元病 (Motor neuron disease,MND)
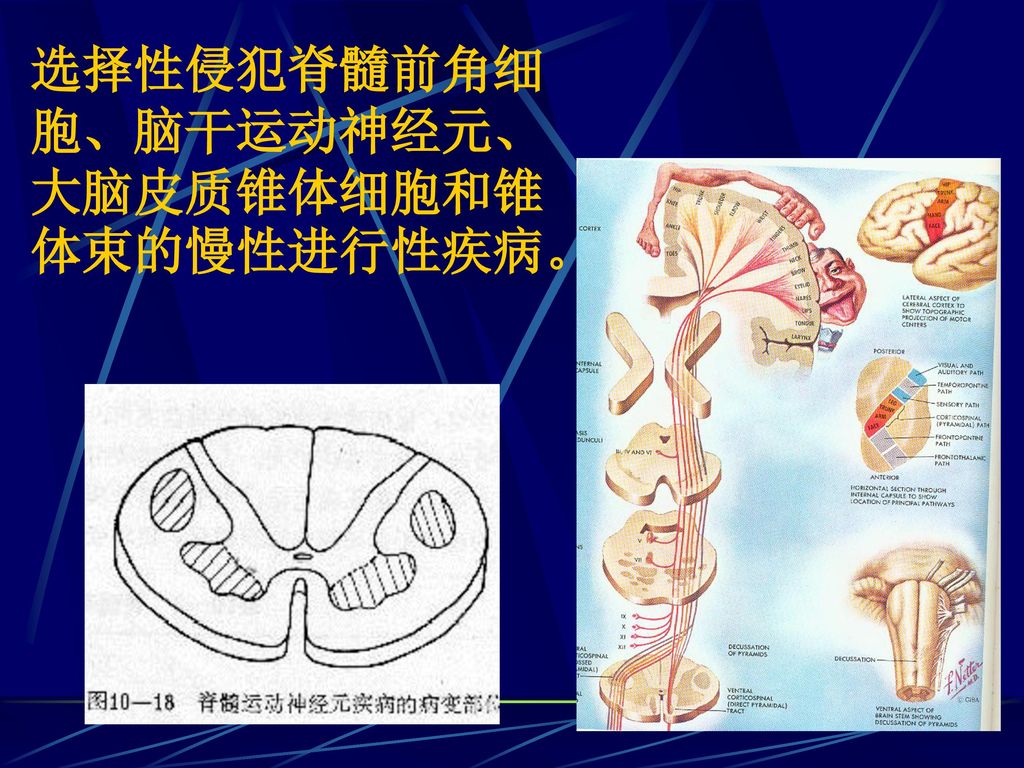
选择性侵犯脊髓前角细胞、脑干运动神经元、大脑皮质锥体细胞和锥体束的慢性进行性疾病。
病因及发病机制 中毒因素 植物毒素、微量元素、神经营养因子减少。 遗传因素 5-10%ALS与遗传有关,多数散发。 免疫因素 尚不明确。 慢病毒感染及恶性肿瘤
病理 神经细胞变性、数目减少。变性细胞深染固缩,胞浆内见脂褐质沉积,星形胶质细胞增生。 病理 神经细胞变性、数目减少。变性细胞深染固缩,胞浆内见脂褐质沉积,星形胶质细胞增生。 大脑皮层运动区锥体细胞 脑干下部运动神经核 脊髓前角细胞
临床表现 一、肌萎缩侧索硬化 ALS 一般为中年后发病,男>女 上、下运动神经元同时受损 延髓麻痹通常晚期出现 无感觉症状,对智力膀胱括约肌等 无影响 病程持续进展,平均3-5年 肌电图有神经元性损害
二、进行性脊肌萎缩症 (Progressive spinal muscular atrophy) 变性限于脊髓前角细胞。 表现为下运动神经元损害症状。 发病年龄在20—50岁之间大多30岁前后。起病缓慢。男多于女。 90%以颈膨大损害开始故以一侧或双侧手肌无力、大小鱼、骨间肌、蚓状肌萎缩,严重者可有“爪形手”。逐渐发展至上肢。首发于下肢者少见。 有肌束颤。 肌张力、反射减退。 病理征阴性,感觉正常。 波及延髓者存活时间短.
三、进行性延髓麻痹 (Progressive bulbar palsy) 临床表现为构音不清、嘶哑、鼻音、咳呛、流 延、吞咽困难。 检查可见软腭运动、咽喉肌无力、咽反射消失舌肌萎缩、舌肌束颤似蚓蠕动。 面肌受累可见表情淡漠、呆板。 双侧皮质延髓束受累:出现强哭、强笑、下颌反射、掌颏反射亢进,噘嘴反射明显等假性球 麻痹症状。 发展迅速1—2年因呼吸肌麻痹、肺部感染死亡。
四、原发性侧索硬化 Primary lateral sclerosis 选择性损害锥体束,极少见,中年以后起病 首发双下肢对称性上运动神经元瘫,渐向上发展,无肌萎缩,感觉正常 皮质延髓束变性出现假性球麻痹 多缓慢进行性
辅助检查 MRI 部分受累脊髓和脑干萎缩 EMG 神经原性改变 肌肉活检 早期为神经原性肌萎缩,无特异性
诊断 中年以后隐袭起病,6个月内逐步进展 延髓、颈、胸、腰骶支配节段中有3个出现上、下运动神经元损害 疾病常由腰骶向颈、胸、延髓发展 肌电图神经元性改变
运动神经元病没有的表现 感觉障碍 明显的括约肌障碍 视觉和眼球运动障碍 自主神经系统及锥体外系障碍 痴呆
鉴别诊断 颈椎病脊髓型 发病年龄、症状相似,胸锁乳突肌肌电图基本正常。 脊髓空洞症 分离性感觉障碍,MRI可见空洞。 颈髓肿瘤 脑干肿瘤
治疗 目前尚无有效治疗 支持治疗 对症治疗 预防肺部感染,人工呼吸 新药 阻断CNS内谷氨酸能神经传递药力鲁唑(riluzole),50mg,Bid口服,可推迟呼吸功能障碍的时间,延长生存期。
思考题 运动神经元病有哪几种类型? 运动神经元病须与哪些疾病鉴别?如何鉴别?
