Download presentation
Presentation is loading. Please wait.

1
抗癌药物的药代动力学

2
药物的药代动力学简介 药物的药代动力学参数意义 抗癌药物的药代动力学参数及其意义

3
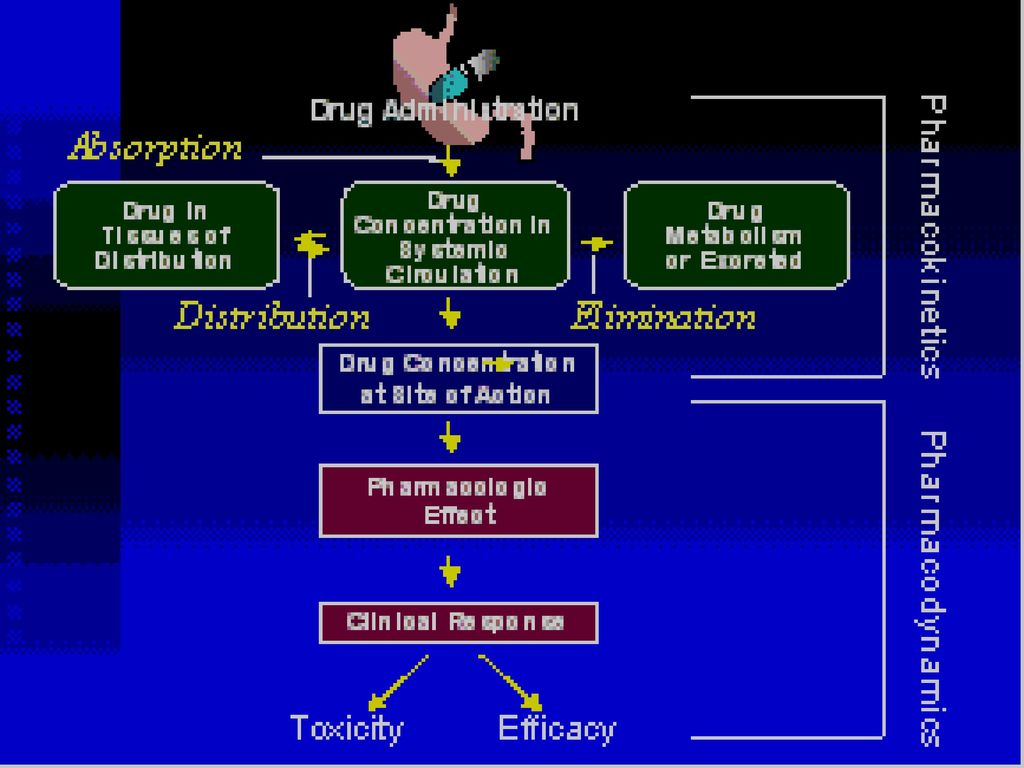
一、药物的体内过程 吸收 排泄 分布 转化 药动学过程 药效学过程 效应 药物

6
ADME系统 指药物体内过程的吸收(Absorption)、分布(Distribution)、代谢(Metabolism)及排泄(Excretion)。可概括为药物的转运和转化。 作用:制定合理的临床给药方案;比较新药的生物等效性的等。 药代动力学: 一门用时间函数来定量描述药物在体内的吸收、 分布、生物转化和排泄过程及其规律的学科。

7
一、药物的体内过程 生 物 转 化 排 泄 分 布 吸 收

8
⒈ 药物跨膜转运

9
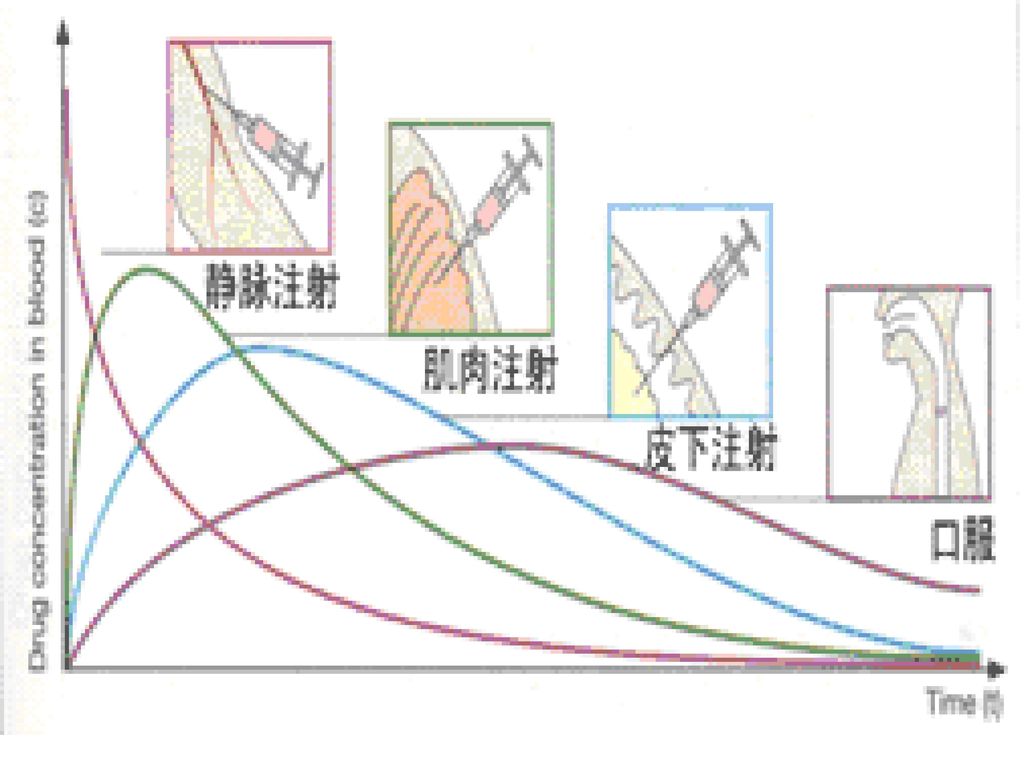
2. 吸 收 ① 概念:从给药部位进入全身循环 ② 分类: →口服给药 (Oral ingestion) *吸收部位主要在小肠 *停留时间长,经绒毛吸收面积大 *毛细血管壁孔道大,血流丰富 *PH5-8,对药物解离影响小
 *吸收部位主要在小肠 *停留时间长,经绒毛吸收面积大 *毛细血管壁孔道大,血流丰富 *PH5-8,对药物解离影响小.")
10
→肌肉注射和皮下注射 *被动扩散+过滤,吸收快而全 *毛细血管壁孔半径40?,大多水溶性药可滤过 → 呼吸道吸入给药
→静脉注射给药 →肌肉注射和皮下注射 *被动扩散+过滤,吸收快而全 *毛细血管壁孔半径40?,大多水溶性药可滤过 → 呼吸道吸入给药 *气体和挥发性药物(全麻药)直接进入肺泡,吸收迅速 *肺泡表面积大( m2) *血流量大(肺毛细血管面积80 m2 ) →经皮给药
直接进入肺泡,吸收迅速 *肺泡表面积大( m2) *血流量大(肺毛细血管面积80 m2 ) →经皮给药.")
11
药物吸收----静脉注射 注射 快速滴注 恒速滴注 起效时间 快 稍慢 较慢 最高浓度 -药物浓度 -滴注速度 -滴注速度 -滴注速度
注射 快速滴注 恒速滴注 起效时间 快 稍慢 较慢 最高浓度 -药物浓度 -滴注速度 滴注速度 -滴注速度 总剂量 特点 血药波动 维持峰浓度 血药浓度波 动小安全性较大

13
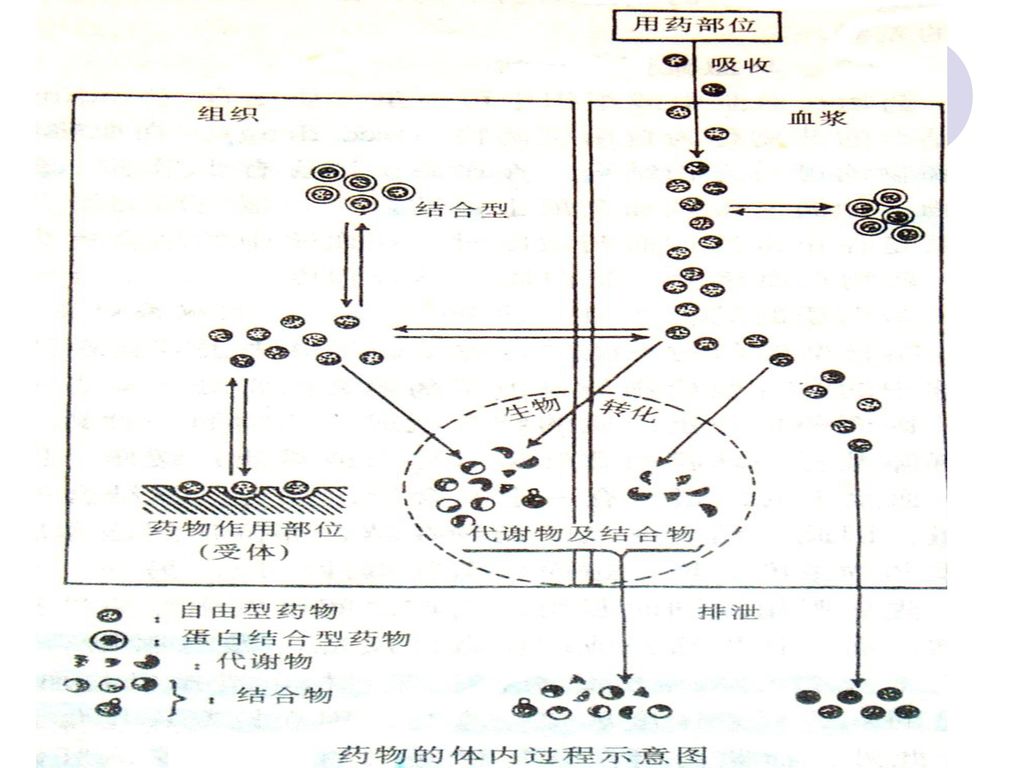
3. 药物分布 药物从血循到达作用、储存、代谢、排泄等部位
*脂溶度 *局部 pH 和药物离解度 *毛细血管通透性 *组织通透性 *转运蛋白量 *血流量和组织大小 *血浆蛋白和组织结合

14
→血浆蛋白结合 影响因素: *可逆性 *可饱和性 *DP不能通过细胞膜 *非特异性和竞争性 →血脑屏障 特点:

15
→胎盘屏障 特点: *胎毛细血管内皮对药物转运的选择性
→胎盘屏障 特点: *胎毛细血管内皮对药物转运的选择性 *脂溶度、分子大小是主要影响因素 (<MW 600易通过;>1000 不能) *母血pH = 7.44; 胎血pH=7.30。弱碱性药物在胎血内易离解 *胎盘有代谢(如氧化)药物的功能 *转运方式和其它细胞相同:简单扩散 *大多数药物均能进入胎儿
 *母血pH = 7.44; 胎血pH=7.30。弱碱性药物在胎血内易离解 *胎盘有代谢(如氧化)药物的功能 *转运方式和其它细胞相同:简单扩散 *大多数药物均能进入胎儿.")
16
4.生物转化 药物作为外源性物质在体内发生化学结构的改变,脂溶性降低,极性增加,易排出体外 器官:肝脏,肠粘膜,肾,肺,体液,血液
前药 活性药物 活性代谢物 非活性代谢物 结合型衍生物

18
5.药物的排泄 药物及其代谢产物经机体的排泄或分泌器官排出体外的过程 器官:肾脏,肺,胆汁,肠道,唾液腺,乳腺,汗腺 特点: ①被动转运
②治疗价值---同时造成不良反应 ③排泄器官功能障碍均能引起排泄速率减慢,药物蓄积

19
①肾排泄---肾小球滤过 ---肾小管分泌 ---肾小管重吸收 药物排泄滤=(1-FR)*(滤过率+分泌率) ②胆汁排泄----主动分泌过程 ③肠道排泄 ④其它途径排泄:唾液,乳汁,汗液和泪液
*(滤过率+分泌率) ②胆汁排泄----主动分泌过程 ③肠道排泄 ④其它途径排泄:唾液,乳汁,汗液和泪液")
20
二、药代动力学参数 定义:由实验得到的时量曲线经数据处理可得到 药物在体内吸收、分布和消除各环节的参数 意义:
①定量描述药物的体内过程和给药后血药浓度的变化规律 ②调节和控制血药浓度以达到期望的药物效应

21
⒈ 房室模型 概念:是药动学研究中按药物在体内转运速率差异,以实验与理论计算结合设置的数学模型 分类: 开放性一室模型 开放性二室模型
无房室模型

22
一室模型VS二室模型 一室模型 二室模型 将整个机体看作一个房室 将机体划分为两个房室 (血流量多、血流速度快的 组织器官构成中央室,其余
一室模型 二室模型 将整个机体看作一个房室 将机体划分为两个房室 (血流量多、血流速度快的 组织器官构成中央室,其余 构成周边室) 机体组织内药量 周边室的药物要返回中央室代 与血浆内药物分子 谢与排泄 瞬时取得平衡
 机体组织内药量 周边室的药物要返回中央室代. 与血浆内药物分子 谢与排泄. 瞬时取得平衡.")
24
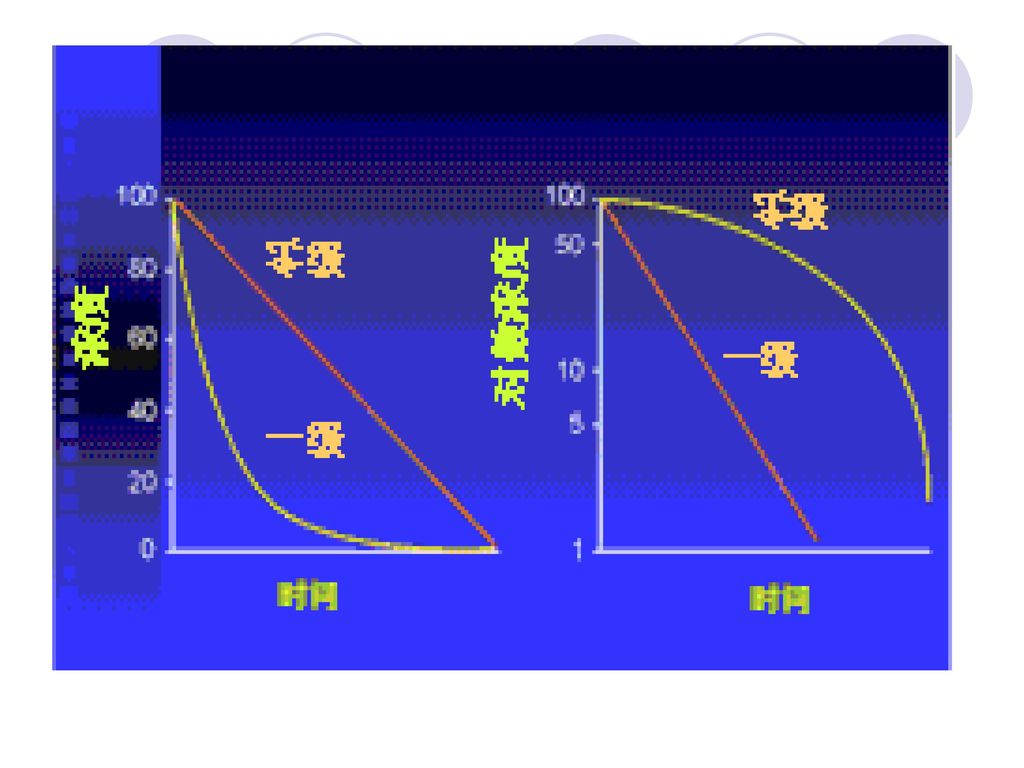
⒉ 半衰期(t1/2) ◆定义: 生物半衰期:药物效应下降一半所需的时间 血浆半衰期:是指药物得血浆浓度下降 一半所需的时间
消除半衰期:是指消除相时血浆药物浓 度降低一半所需的时间

25
反映机体消除药物的能力与消除药物的快慢程度
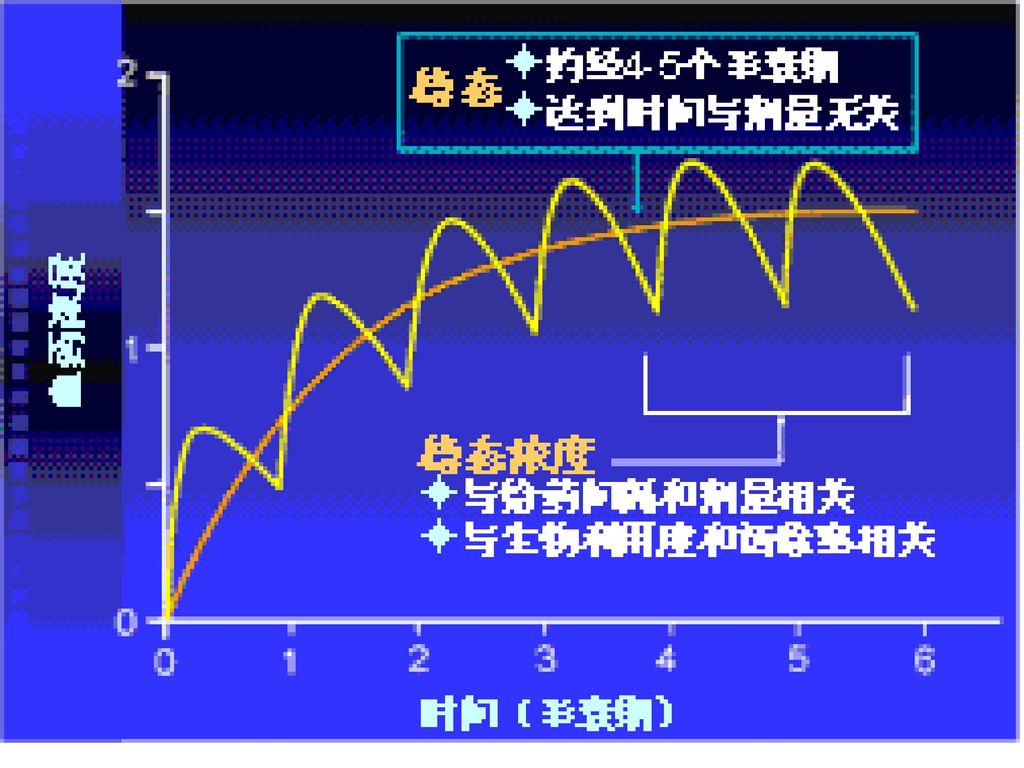
◆意义: 反映机体消除药物的能力与消除药物的快慢程度 与药物转运和转化关系行为,单次给药后,经过5~6个t1/2 ,体内药物消除基本干净(消除96.9%);每间隔一个t1/2用药1次,则给药5次后,血药浓度达稳态血药浓度 按t1/2长短将药物分为5类: 超短效(t1/2≦1h) 短效(t1/2 1~8h) 中效 (t1/2 4~8h) 长效(t1/2 8~24h) 超长效(t1/2>24h)
;每间隔一个t1/2用药1次,则给药5次后,血药浓度达稳态血药浓度. 按t1/2长短将药物分为5类: 超短效(t1/2≦1h) 短效(t1/2 1~8h) 中效 (t1/2 4~8h) 长效(t1/2 8~24h) 超长效(t1/2>24h)")
26
⒊ 表观分布容积(Vd ,L.kg-1 ) ◆定义:假设药物均匀的分布于各种组织与 体液,且其浓度与血液种相同,在这种假想
条件下药物分布所需的容积。 ◆是个数学概念,并不代表具体的生理空间 ◆代表给药剂量或体内的药物总量与血浆药 浓度相互关系得一个比例常数Vd=Dose/Css

27
④意义 ◆估算血容量及体液量 ◆反应药物分布的广度和药物与组织结合的程度 。 正常体液值:0.6L/kg 药物Vd为0.1~0.3L/kg,表明药物不易进入组织 药物Vd>0.6L/kg,表明有组织蓄积

28
◆代表药物透膜转运和分布到体内各部位的特性。
除了蛋白结合率极高的药物,分布容积小的药物排泄越快,体内存留时间短;分布容积大的药物排泄越慢,在体内存留时间越长 ◆根据药物的分布容积调整剂量 D=Vd *C ◆对于抗癌药物特别是作用于实体瘤的药物,Vd大可能与其抗肿瘤药物的疗效有关。

29
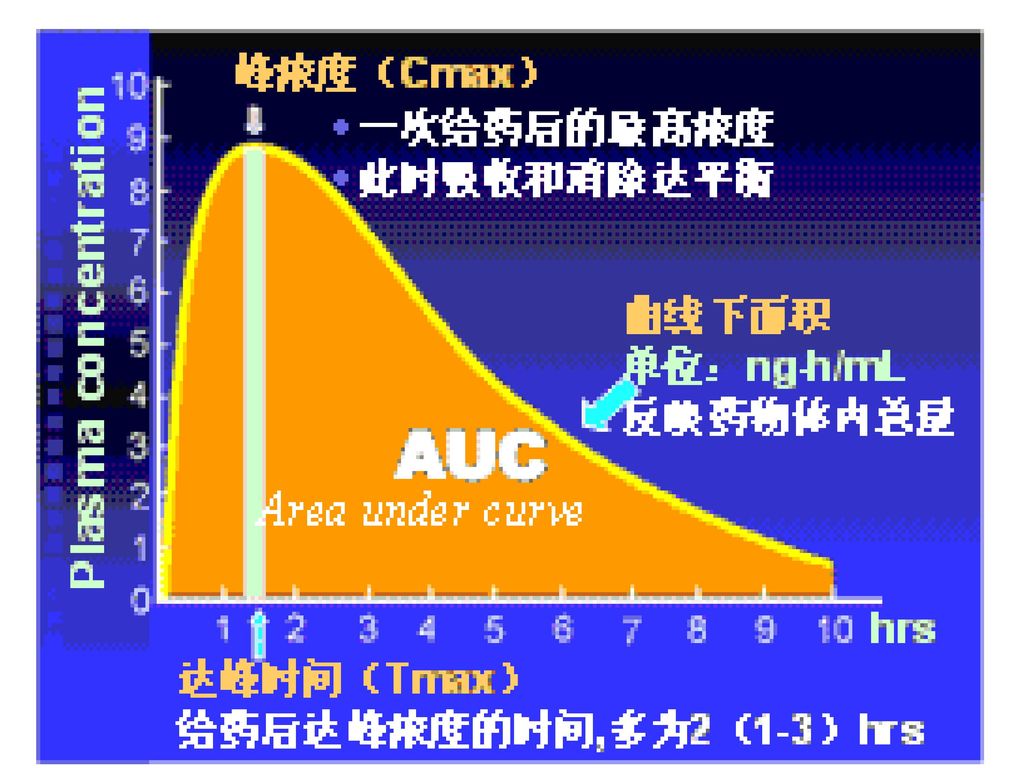
⒋ 血药浓度-时间曲线下面积(AUC) ①定义:以血浆药物浓度为纵坐标,时间为横坐标,绘出的曲线为血药浓度-时间曲线,坐标轴和血药浓度-时间曲线之间所围成的面积称为AUC。 ②意义:许多抗癌药物的疗效和毒性都与AUC有关
 ①定义:以血浆药物浓度为纵坐标,时间为横坐标,绘出的曲线为血药浓度-时间曲线,坐标轴和血药浓度-时间曲线之间所围成的面积称为AUC。 ②意义:许多抗癌药物的疗效和毒性都与AUC有关.")
31
5. 稳态及稳态动力学参数 ◆定义:在恒定给药间隔时间重复给药,当一个给药间隔内的摄入药量等于排出量时,血药浓度达到稳态。 ◆所有的药物到达稳态的时间只与药物本身的半衰期有关,一般给药后4~5个半衰期到达稳态 ◆稳态的药-时曲线

32
◆稳态的药代动力学参数 稳态血药浓度:Css 最高稳态血药浓度:(Css)max 最低稳态血药浓度:(Css)min 平均稳态血药浓度 积累系数:R 负荷剂量:DL
max 最低稳态血药浓度:(Css)min 平均稳态血药浓度 积累系数:R 负荷剂量:DL")
34
6. 清除率(CL) ◆定义:是指单位时间内整个机体和某消除器官能消除多少毫升血中所含的药物,即单位时间消除的药物表观分布容积。 CL=CLH+CLR
 ◆定义:是指单位时间内整个机体和某消除器官能消除多少毫升血中所含的药物,即单位时间消除的药物表观分布容积。 CL=CLH+CLR")
35
Cockcroft&Gault ◆肾清除率:包括肾小球滤过、肾小管细胞主动分泌和重吸收。
若药物只是经肾小球滤过,正常CL大约为125ml/min Cockcroft&Gault 男性:CLcr(ml/min)=(140-年龄)体重(kg) 72 血清肌酐(mg/dl)女性:CLcr(ml/min)=0.85 (140-年龄)体重(kg) 72 血清肌酐 (mg/dl)
=(140-年龄)体重(kg) 72 血清肌酐(mg/dl)女性:CLcr(ml/min)=0.85 (140-年龄)体重(kg) 72 血清肌酐 (mg/dl)")
36
如果发生药物再吸收,清除率在1~125ml/min,如果99%的药物被再吸收,则清除率接近于1ml/min。
影响因素:尿液PH、血浆蛋白结合程度以及肾血流量。

37
◆肝清除率(CLH) CLH=QH [ fu CLint ] QH+fu.Clint QH:肝血流量,fu:血液中未结合药物分数 Clint:肝脏总的内在清除活性 影响因素:肝血流量、药物在血浆内的结合及肝酶的内在活性,药物在肝血窦中被摄取,药物被代谢和/或胆汁排泄消除
![◆肝清除率(CLH) CLH=QH [ fu CLint ] QH+fu.Clint. QH:肝血流量,fu:血液中未结合药物分数.](http://slidesplayer.com/slide/11360659/61/images/37/%E2%97%86%E8%82%9D%E6%B8%85%E9%99%A4%E7%8E%87%EF%BC%88CLH%EF%BC%89+CLH%3DQH+%5B+fu+CLint+%5D+QH%2Bfu.Clint.+QH%3A%E8%82%9D%E8%A1%80%E6%B5%81%E9%87%8F%EF%BC%8Cfu%EF%BC%9A%E8%A1%80%E6%B6%B2%E4%B8%AD%E6%9C%AA%E7%BB%93%E5%90%88%E8%8D%AF%E7%89%A9%E5%88%86%E6%95%B0..jpg "Clint:肝脏总的内在清除活性. 影响因素:肝血流量、药物在血浆内的结合及肝酶的内在活性,药物在肝血窦中被摄取,药物被代谢和/或胆汁排泄消除.")
38
7. 速率常数(k) ◆定义:可定量的比较药物转运速率的快慢,速率常数越大,过程越快 ◆K:一级消除速率常数 ◆K12:二房室模型药物从中央室进入周边室的速率常数 ◆K21:二房室模型药物从周边室进入中央室的速率常数

39
⒏ 生物利用度(F) ◆定义:指药物吸收进入血循环的程度和速率,指药物吸收进入血液循环的速度和程度
◆意义:评价药物制剂质量得重要指标,也是选择给药途径得依据之一 ◆分类:相对生物利用度(F) 绝对生物利用度(Fr)
 绝对生物利用度(Fr)")
40
小 结 在所有的药代动力学参数中,CL和Vd与剂量无关,只与病人本身的肝、肾功能,血浆蛋白清除率,血浆和尿的PH有关。
小 结 在所有的药代动力学参数中,CL和Vd与剂量无关,只与病人本身的肝、肾功能,血浆蛋白清除率,血浆和尿的PH有关。 可以通过AUC和Css调节剂量,从而达到目 标AUC和Css。 Dose=AUCCL Dose=Css Vd

41
抗癌药物药代动力学参数及其应用 烷化剂 抗代谢类 抗生素类 植物类 铂类 其它类

42
烷化剂 ⒈ 机制:C+集电集团进攻 DNA,RNA,蛋白质富电子位点,引进烷基,即用自身的烷基取代生 物大分子的氢 ⒉分类 脂肪氮:氮芥
①氮芥类 芳香氮 RN-CH2CH2CL 杂环氮 甾体氮芥 ②亚硝酸类 ③乙亚胺类

43
F 药动学 分布 盐酸氮芥 0 t1/2 <1m 48min降低65%-85% 肺、小肠、 (恩比兰) 肾、脾、肌 肉、脑中最少
(恩比兰) 肾、脾、肌 肉、脑中最少 硝卡芥 % t1/2 较长 24h减少54% 胆囊、肾最多, (消瘤芥) Iv1h后已广泛分布全身组织 脑中最少,但 通过血脑屏障 肝、肾最高 甘磷酰芥 口服,8h达峰,维持8hh 盐酸氧氮芥(癌可平) 同恩比兰 美法仑 吸收不完全, h 需调整剂量, 蛋白结合率<30%, 轻 度白细胞减少 个体差异大
 肾、脾、肌. 肉、脑中最少. 硝卡芥 78% t1/2 较长 24h减少54% 胆囊、肾最多, (消瘤芥) Iv1h后已广泛分布全身组织 脑中最少,但. 通过血脑屏障. 肝、肾最高. 甘磷酰芥 口服,8h达峰,维持8hh. 盐酸氧氮芥(癌可平) 同恩比兰. 美法仑 吸收不完全, 1-2h 需调整剂量, 蛋白结合率<30%, 轻 度白细胞减少. 个体差异大.")
44
抗瘤新芥 口服吸收缓慢,6h达峰 min (邻脂苯芥) N-甲酰溶肉瘤素 口服吸收迅速 小时迅速消失 (氯甲) 小时达峰 甲氧芳芥 口服吸收迅速; h 骨髓、肾、 (甲氧基溶肉瘤素) 有一定的蓄积作用; 肝脏 不宜大剂量连续用药 苯丁酸氮芥 口服迅速完全,达峰时间40-79min (瘤可宁) 代谢无抗瘤作用,显效慢,2-6周才出现 间歇给药比每日小剂量维持 给药对骨髓的毒性
 有一定的蓄积作用; 肝脏. 不宜大剂量连续用药. 苯丁酸氮芥 口服迅速完全,达峰时间40-79min. (瘤可宁) 代谢无抗瘤作用,显效慢,2-6周才出现. 间歇给药比每日小剂量维持 给药对骨髓的毒性.")
45
◆年龄>70y的患者,骨髓抑制发生率增加
环磷酰胺 机制:体外无活性,进入体内被肝脏或肿瘤内存在的磷 酰胺酶或磷酸酶水解,变为活化型的磷酰胺氮芥 药代动力学特点: ◆口服易吸收,达峰时间为1h,t1/2为4~6h,48h经肾脏排泄50~70%,血浆蛋白结合率50% ◆70%经肝脏代谢:中度肝功能损害---暴露在药物活性并没有改变,不需调整剂量 ◆相互作用 西咪替叮---提高4-OH CTX血药浓度 吗啡、泼泥松龙---抑制CTX代谢转化 ◆年龄>70y的患者,骨髓抑制发生率增加

46
◆注意事项: 代谢产物对尿路刺激性 提高剂量强度,能明显增加疗效,非血液学毒性增加,心肌炎,中毒性肝炎及肺纤维化 腔内给药无直接作用
水溶液稳定性较差,现配现用

47
异环磷酰胺 药代动力学特点: ◆年龄>60y t1/2 6.03h < 60y t1/2 3.85h
◆代谢物主要经肾脏排泄 CLCR60ml/min % CLCR45ml/min % CLCR30ml/min % ◆异环磷酰胺剂量分开几天给药,或持续滴注可提高治疗指数

48
◆机制:抑制二氢叶酸还原酶r为与一个或多个麸氢酸结合,具有强大的抑制作用,并且荷负电荷不易逸出细胞,产生持久效应
抗代谢类 氨甲喋啉(MTX) ◆机制:抑制二氢叶酸还原酶r为与一个或多个麸氢酸结合,具有强大的抑制作用,并且荷负电荷不易逸出细胞,产生持久效应 ◆剂型:口服 0.1mg/kg 完全吸收 >25mg/kg 口服吸收饱和 >10mg/kg 口服吸收1/10
 ◆机制:抑制二氢叶酸还原酶r为与一个或多个麸氢酸结合,具有强大的抑制作用,并且荷负电荷不易逸出细胞,产生持久效应. ◆剂型:口服 0.1mg/kg 完全吸收. >25mg/kg 口服吸收饱和. >10mg/kg 口服吸收1/10.")
49
◆药代动力学特点 体内过程: t1/2 2~8min t1/2 2~3h t1/2 8~10h 蛋白结合率 60% 主要经肾脏排泄,肾小球滤过,肾小管分泌排 泄,肌肝消除率<60ml/min 不予使用MTX

50
相互:与下面药物合用将减少减少肾脏对其的排
泄 probenecid 丙磺舒 Penidlins Cephalosporins Aspirin 肝脏代谢为7-OH-MTX,胆汁排泄10%胸水、 腹水末端消除半衰期延长,毒性增加 CL急性淋巴白血病复发比率

51
水化 High-dose lymphomas Osteogenic sarcoma 6~42h Acuife leukemia 碱化尿液
水化 碱化尿液 监测MTX浓度 足够给予LV

52
LV的调整 48h MTX 0.5mm 15mg/ m2. 1.0mm 100mg/ m2. 2.0mm 200mg/ m2.
>10mm LV很难解毒 35~40ml/min LV过多-----解救肿瘤细胞 过少-----毒性作用(静脉给药) 口服 剂量>40mg. 生物利用度
 口服 剂量>40mg. 生物利用度")
53
氟尿嘧啶 ◆机制:在体内经尿苷磷酸激酶转化为5-氟脱氧尿 嘧啶核苷酸,通过抑制胸腺嘧啶核苷酸合成酶而 抑制DNA合成 ◆药代动力学特点:
口服吸收无规则,个体差异大,肠道粘膜存在高水平的DPD酶。 t1/2为8-14min, 85%由DPD酶代谢 (肝、淋巴细胞、肠道) 3%-5%人缺乏DPD酶(外显子突变,缺失)。 Vd大、 CSF、第三室(胸水、腹水)。
 3%-5%人缺乏DPD酶(外显子突变,缺失)。 Vd大、 CSF、第三室(胸水、腹水)。")
54
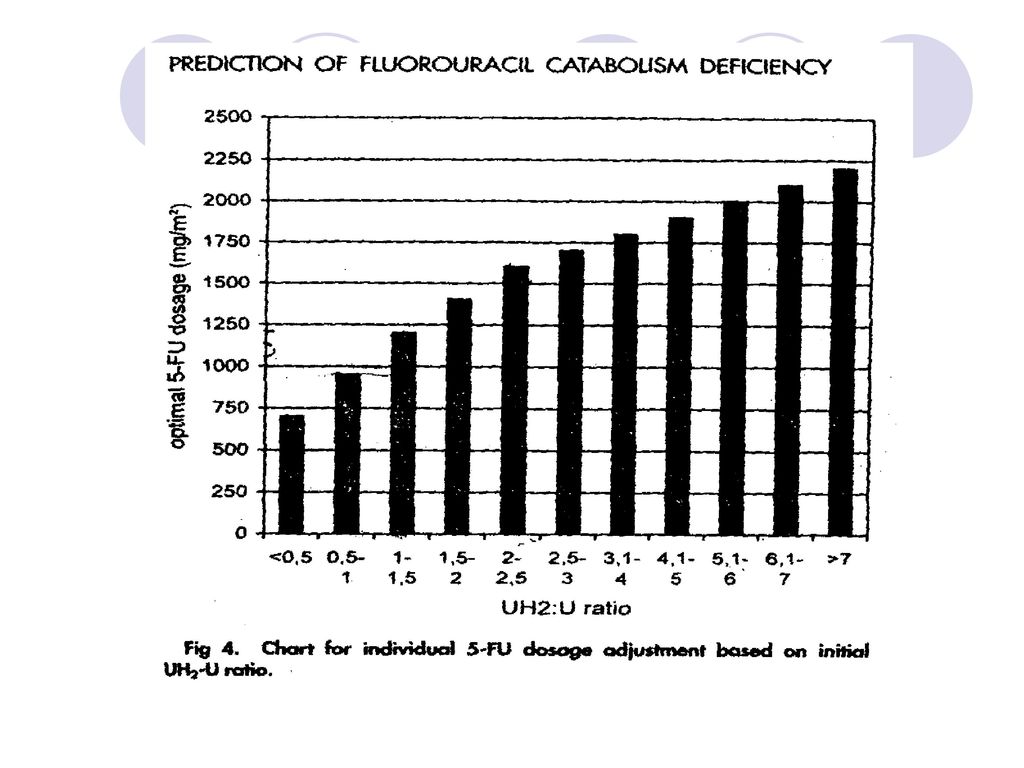
◆测定:双抗体夹心法,放射-HPLC法 测定H2U/U间接反应DPD酶的活性 ◆正常值:4.5>H2U/U >2.25 部分缺失:20% 中度缺失 :50% 重度缺失:100% Gamelin 等人研究每周给药方案,8h恒速静脉滴注,探讨了根据DPD酶活性调节剂量。 治疗窗:Css:2000~3000ug/L AUC:16~24mg/L.h

55
◆给药方式:静推,持续静脉滴注 静推 : mg/m2 Cmax :300m-1m 2h后血药浓度低 于1m 5-Fu消除只有饱和,剂量增加,非线性增加 持续静脉滴注:5000mg/ m2 毒性与AUC有关,通过调整 Css,即调整滴速,来避免严 重的毒 付反应。

57
氟尿苷 机制:快速静脉注射在体内被迅速代谢失活,而在缓 慢动脉灌注时,可在体内转化为活化型氟尿苷单磷酸 盐,抑制TP酶 5-FU FudR
Fudr mg/kg/d 连用14天,小剂量持续灌注患 者好耐受,给药方式不同毒性不一样

58
卡培他滨 ◆机制: 羧酸酯酶 Xxloda ’-DFCR 胞苷脱胺酶 5’-DFUR 胸苷磷酸化酶 5’-FU

59
◆药代动力学特点: 生物利用度:80%,食物会降低吸收速率和吸收程度 肿瘤组织胸苷磷酸化酶是正常组织的3.5倍 原形药物1.5h达峰,代谢物5-Fu 2h达峰 肿瘤组织浓度是血浆浓度的20倍 T1/2为3~4h DPD酶缺乏患者慎用

60
阿糖胞苷 ◆机制:经磷酸激酶转化为阿糖胞苷三磷酸及阿糖胞苷而磷酸,抑制DNA 聚合酶及抑制二磷酸胞苷转变为二磷酸脱氧胞苷 药代动力学特点:
口服吸收少且不规则,极易被肠道粘膜及肝脏 的胞嘧啶核苷酸失活

61
小剂量 100mg-200 mg/m2 /d mmol CSF50%血药浓度 缺胞苷脱氨酶,半衰期2-4h 大剂量 Ara-c >2 g/m h 滴注 α 16min β 1.8h γ 6h 血药浓度60-150mm,12h后浓度<0.5 mm CSF50%血药浓度:10mmol

62
基因多态性 CDK---脱氧胞苷激酶 SNP rs1780842 G1T1----ERK1/2 RAD51AP1 CDA---胞苷脱氨酶
NT5CZ---5’-核苷酸酶 HENT1---核苷酸转移因子

63
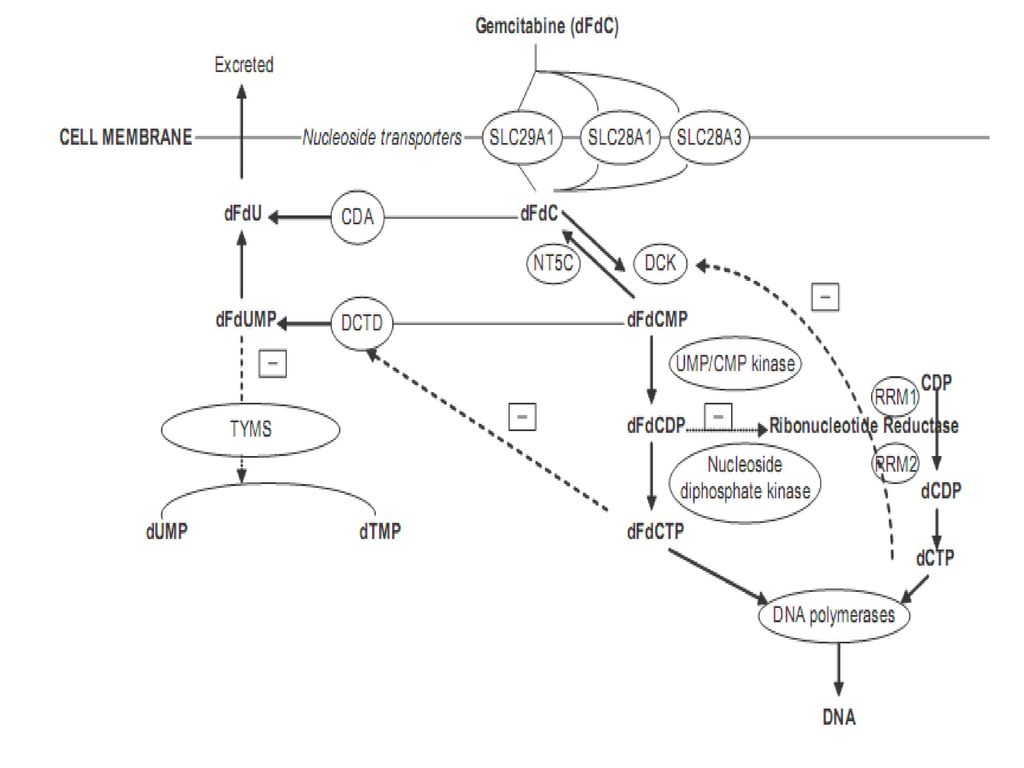
吉西他滨 ◆机制: dFdCDP---抑制 核糖核苷酸还原酶 dFdCTP---可与dCTP竞争性结合到DNA上,干 扰DNA的合成
与Ara-C相比,Gem的磷酸化效率强6倍,且不易脱氨排泄

64
PK参数 dFdC 8 min dFdU t1/2 27 min; t1/2 14 h
dFdCTP t1/2 3.9 h; t1/2 16 h CL:30倍的个体差异

66
核酸转移酶 SLC29A1 Mrn DCK 缺乏---耐药 特点:随着年龄↑,tumor DCK表达↓ OS、PFS ↓ 360G/201-T 高的效率

67
◆ 活化酶:deoxycytidien kinase (DCK)
失活酶:cytidine deaminase (CD) deoxycytidylate deaminase (DCD) %decrease WBC ANC Paltelets CD< ± ± ±18.9 CD> ± ± ±20.6 P
 deoxycytidylate deaminase (DCD) %decrease WBC ANC Paltelets. CD< ± ± ±18.9. CD> ± ± ±20.6. P")
68
木脂体类 ◆机制:抑制DNA拓扑异构酶II,阻止DNA双链 的重新连接,诱导DNA链断裂 杀伤细胞作用为时间与浓度依赖性

69
VM-26 分子量 588.6 Pka 9.7 44%-60%由肾脏排泄 16%粪便排泄 1%-10%给药后(2-20h)
656.7 10.1 主要经肝代谢 脑脊液浓度<1% 口服吸收非线性,剂量与AUC不成比例 t1/ 2 6~20h 99% VP-16 分子量 Pka 44%-60%由肾脏排泄 16%粪便排泄 1%-10%给药后(2-20h) 静t1/2α h h 口服 t1/ h t1/2 β h 蛋白结合率 %(白蛋白) 低蛋白血症,增加VP-16 的游离血药浓度
 静t1/2α h h. 口服 t1/ h. t1/2 β h. 蛋白结合率 92-94%(白蛋白) 低蛋白血症,增加VP-16. 的游离血药浓度.")
70
VP VM-26 生物利用度 50% % 24~137% ~53% 20~70% ~14% 达峰时间 ~4.0 h ~1.5h 剂量线性 nonlinear F=0.76 at 100mg/m F=0.36 at 100mg/m2 F=0.48 at 100mg/m F=0.24 at 100mg/m2 口服比静脉给药VP16个体差异大

71
VP-16 ①白蛋白↓,胆红素高→游离药物浓度↑,毒性↑ ②游离药物%=1.4×胆红素[mg/dL]
简单研究:血浆白蛋白<35g/L,剂量30~40%↓ respording patiend (24h)>8m <8m (代谢快) ③给药方式与疗效有关(浓度1mg/mL) 500mg/ m2. 24h infusion 46h 50% 2h分5day 95h %
![VP-16 ①白蛋白↓,胆红素高→游离药物浓度↑,毒性↑ ②游离药物%=1.4×胆红素[mg/dL]](http://slidesplayer.com/slide/11360659/61/images/71/VP-16+%E2%91%A0%E7%99%BD%E8%9B%8B%E7%99%BD%E2%86%93%EF%BC%8C%E8%83%86%E7%BA%A2%E7%B4%A0%E9%AB%98%E2%86%92%E6%B8%B8%E7%A6%BB%E8%8D%AF%E7%89%A9%E6%B5%93%E5%BA%A6%E2%86%91%EF%BC%8C%E6%AF%92%E6%80%A7%E2%86%91+%E2%91%A1%E6%B8%B8%E7%A6%BB%E8%8D%AF%E7%89%A9%EF%BC%85%EF%BC%9D1.4%C3%97%E8%83%86%E7%BA%A2%E7%B4%A0%5Bmg%2FdL%5D.jpg "简单研究:血浆白蛋白<35g/L,剂量30~40%↓ respording patiend (24h)>8m. <8m (代谢快) ③给药方式与疗效有关(浓度1mg/mL) 500mg/ m2. 24h infusion 46h 50% 2h分5day 95h 89%")
72
30~50% 经肾脏排泄 CLCR60ml/min % CLCR45ml/min % CLCR30ml/min % 肝脏对其代谢不很清楚,肝功能损害,并不产生肾对其代谢等率的增加而弥补。 与异环磷酰胺合用,CL↑28% 24h血药浓度>0.33 mg/L ,中性粒细胞↓50%

73
Excretion Metaboliam Not bound to protein Articipatid effect (Hepatic)
× × - × - × × + × - × + - × + ± × - + ± - - + +

74
蒽环类抗生素 特点:色素苷元 氨基糖 侧链 机制:插入DNA 抑制TopoII Fe2+-ADM-DNA

75
Doxorubicin Daunorubicin Epirubicin Idarubicin
F NA NA NA ~45% Vdss 214~ ~ ~ ~2556 P U 15~ NA T1/2 0.43~ ~ ~ ~6.0 T1/2 20.6~ ~ ~ ~42.7 CL ~ ~ ~ ~122.6 MT less active inactive low cytotoxicity cytotoxic EL bile and urinary

76
主要毒性 骨髓抑制 心脏毒性 其它毒性 脱发,口腔炎,恶心,呕吐 相互作用 多功能氧化酶诱导剂如苯巴 比妥钠可增加心脏毒性 与肝素等产生沉淀

77
Doxorubcicin(阿霉素) 70%与血浆蛋白结合 血浆蛋白↓ →调整剂量 肝功能损害→AUC↑→肾髓抑制↑→剂量调整
70%与血浆蛋白结合 血浆蛋白↓ →调整剂量 肝功能损害→AUC↑→肾髓抑制↑→剂量调整 胆红素 2~3 mg/dL % >3 mg/dL % >5mg/dL 不使用 个体差异大,胆红素>3 mg/dL ,首个疗程的AUC,测定,个体化疗药。

78
Idarubicin(去甲柔红霉素) 血清肌酐>=2mg/dL 剂量75% 肝脏 中度,高度损害----剂量调整 总胆红素 1.5~5.0 mg/dL AST 60~180 U/L % 总胆红素 > - Epirubicin(吡喃阿霉素) 经肝脏代谢 CL→AST有关与胆红素,碱性磷酸酯酶、蛋白、肌酐无关 AST→预示吡喃阿霉素的药代动力学行为
 经肝脏代谢. CL→AST有关与胆红素,碱性磷酸酯酶、蛋白、肌酐无关. AST→预示吡喃阿霉素的药代动力学行为.")
79
⒈机制:微管-微管蛋白N-末端31氨基酸 ⒉生物利用度低 p-gp 紫杉醇类 可逆的 最低有效浓度 1mol(泰素)
首过效应

80
Paclitaxel Docetaxel 剂量 (mg/m2) (24h),175~225(3h) ~100 药代行为 分布/消除饱和 三相 T1/2 ~20h ~20h CL ~25L/h L/h 代谢 肝代谢,胆道消除 肝代谢 ,胆道消除 代谢酶 CYP2C CYP3A4 主要毒性 白细胞下降 白细胞下降 其它毒性 脱发,神经毒性 脱发,皮肤毒性 肌痛,过敏反应,无 力 无力,肌痛, 神经毒性

81
Paclitaxel(泰素) 药代动力学特点: ◆蛋白结合率 97%, 71%由粪便排泄,14%由肾脏排泄 ◆调整剂量 白蛋白过少症
◆调整剂量 白蛋白过少症 肝功能损害 ◆AST不超过正常值的2倍 胆红素≦1.5mg/dL mg/m2。 1.6≦ 胆红素 ≦3.0mg/dL 75 mg/m2 胆红素 >3.0mg/dL mg/m2 。

82
◆ <125mg/m2. over 3 hours 细胞分布达到饱和
剂量血药浓度不成比例 ◆药物相互作用 T followed P CL 减少 33% 白细胞下降 A followed P A CL 减少 33% 心脏毒性增加 机制:竞争p-gp, oil vehicle ,代谢酶

83
C+P VS C 白细胞下降(-),血小板下 降减少 P followed CTX VS CTX followed P 血液学毒性大 诱导嘧啶磷酸化酶活性,增加希洛达的转化 P450诱导剂增加CLp 皮质酮增加Paclitaxel毒性,Amiforstine保护组织, 不影响药物疗效

84
紫杉醇血药浓度与疗效和毒性关系 P t c > CR h PR h PD h P t c > > 61.4 h 疾病进展时间 w <61.4 h w

85
白蛋白包裹的紫杉醇 Cmax (ng/ml) 10255.47±1550.70 4389.57±1315.98 0.0002 Yz
参数 凯素 (260mg/m2/iv 30min) N = 12 紫杉醇注射液 (175mg/m2/iv 3h) N = 8 P value Cmax (ng/ml) ± ± 0.0002 Yz 0.044±0.012 0.067±0.036 0.0585 T1/2 (h) 16.93±4.89 12.13±4.48 0.064 AUC0-t (ng/h.mL) ± ± 0.2472 AUC0-∞(ng/h.mL) (ng/h.mL) (ng/h.mL) ((ng/h.mL) (ng/h.mL) (ng/h.mL) (ng/h.mL) ± ± AUC0-t /D(ng/h.mL) 31.73±5.68 49.22±11.59 0.0009 AUC0-∞/D(ng/h.mL) (ng/h.mL) (ng/h.mL) ((ng/h.mL) (ng/h.mL) (ng/h.mL) (ng/h.mL) 32.45±5.72 50.22±11.41 Vd (L/m2) 529.54±188.06 232.43±84.46 CL (L/h.m2) 21.70±4.32 13.46±2.68 0.0007 MRT0-t (h) 6.73±1.11 6.66±1.60 0.9078 MRT0-∞ (h) 8.11±1.32 7.46±1.79 0.4875
 N = 12. 紫杉醇注射液. (175mg/m2/iv 3h) N = 8. P value. Cmax (ng/ml) ± ± Yz ± ± T1/2 (h) 16.93± ± AUC0-t (ng/h.mL) ± ± AUC0-∞(ng/h.mL) (ng/h.mL) (ng/h.mL) ((ng/h.mL) (ng/h.mL) (ng/h.mL) (ng/h.mL) ± ± AUC0-t /D(ng/h.mL) 31.73± ± AUC0-∞/D(ng/h.mL) (ng/h.mL) (ng/h.mL) ((ng/h.mL) (ng/h.mL) (ng/h.mL) (ng/h.mL) 32.45± ± Vd (L/m2) ± ± CL (L/h.m2) 21.70± ± MRT0-t (h) 6.73± ± MRT0-∞ (h) 8.11± ±")
86
长春生物碱类 机制:微管蛋白 高亲合力结合位点(16~17), kd 1~2mol,位于 微管尾端,抑制微管变长,变短。
V浓度<2 mol 低亲合力结合位点(2), kd 3mol,抑制微管的解聚 特点 :易被细胞摄取,在细胞内蓄积(5~500倍),脂溶性越高,蓄积越多(能量/温度依赖) PK 特点:三室模型,V大
, kd 3mol,抑制微管的解聚. 特点 :易被细胞摄取,在细胞内蓄积(5~500倍),脂溶性越高,蓄积越多(能量/温度依赖) PK 特点:三室模型,V大.")
87
Vincristine Vinblastine Vindesine Vinorelbine
药代动力学模型 三室 三室 三室 三室 半衰期(min) < < < <5 (min)50~ ~ ~ ~168 (h) 23~ ~ ~ ~49 CL `1.29 肝代谢, 胆汁排泄
 <5 <5 <5 <5. (min)50~155 53~99 55~99 49~168. (h) 23~85 20~64 20~24 18~49. CL `1.29. 肝代谢, 胆汁排泄.")
88
Vincristinc (长春新碱) 肝功能损害 AUC↑~3倍 胆红素1.5~3.0mg/dL 剂量50%↓
Nifedipine显著减少长春新碱的清除率,增加AUC

89
长春瑞宾(Oinorelbine) 血小板结合率--- 78% 血小板减少→游离药物↑→血液学毒性↑
胆红素 2.0~3.0mg/dL 50% ↓ > 3.0mg/dL % ↓

90
喜树碱类 机制:选择性的抑制拓扑异构酶I, 能与托扑异构酶及切口的DNA结合形成复合物,引起DNA双链断裂,复制停止。

91
Topotecan CPT-11(SN-38) 50%-65%经肾脏排泄 排泄 胆道排泄
N-desmethy1 topotecan 显有肿瘤活性 0.5% topotecan 5% 尿排泄 肝功能异常毒性、处置topotecan未见改变,不用进行剂量调整。 肾中重度肾功能损害患者 NTD 0.5 µg/m2 /d (5d) CPT-11(SN-38) 排泄 胆道排泄 SN-38-C10 UGT1A1 glucuronic acid 无活性产物,转化程度与腹泻显负相关 SN-38由CMOAT经胆道排泄 肾脏排泄占的部分很小>总胆红素3倍的患者 剂量是常规1/3剂量
 CPT-11(SN-38) 排泄 胆道排泄. SN-38-C10 UGT1A1 glucuronic acid 无活性产物,转化程度与腹泻显负相关. SN-38由CMOAT经胆道排泄. 肾脏排泄占的部分很小>总胆红素3倍的患者. 剂量是常规1/3剂量.")
92
Tapotecan Irinotecan F NA Vdss (< /m2 ) Lactore Total Percent unbound t1/2b(h) Lactoer T.tal CL(L/hr/m) L Total
 Lactore Total Percent unbound t1/2b(h) Lactoer T.tal CL(L/hr/m) L Total")
93
毒性与疗效与AUC相关 毒性:腹泻15%,骨髓16%抑制 合用抗anticovulsants(phonytoin)
腹泻,早性腹泻,滴注早期期间,伴有腹胶痛及面红。 迟发性,用药后数天(DLT) 羧酸酯酶抑制剂 Enkephlinase 抑制剂 抑制SN-38胆道排泄 诱导SN-38 glucuronic acid化 合用抗anticovulsants(phonytoin) 药物topotecan.N代谢增加,可能要增加剂量才能达到同样的AUC 毒性:白细胞下降 d
 羧酸酯酶抑制剂. Enkephlinase 抑制剂. 抑制SN-38胆道排泄. 诱导SN-38 glucuronic acid化. 合用抗anticovulsants(phonytoin) 药物topotecan.N代谢增加,可能要增加剂量才能达到同样的AUC. 毒性:白细胞下降 8-10d.")
94
◆活性低药物,经羧酸酯酶转化为SN-38活性高的药物,转化率有关 3%~15% CPT-11 SN-38 SN-38G
smokers Nonsmokers ◆体内过程: CPT SN-38 65% % ◆CPT-11有一部分与红细胞结合 红细胞结合/血浆蛋白 结合率 与红细胞结合有利于药物转运至肿瘤组织发挥疗效 ◆肝功能损害的患者,AUC增加,毒副反应也增加

95
Topotecan ◆蛋白结合率 35%,血浆蛋白低---影响不大 ◆肾排泄率约占40% 中度肾功能损害需调整剂量
◆蛋白结合率 35%,血浆蛋白低---影响不大 ◆肾排泄率约占40% 中度肾功能损害需调整剂量 1.5 mg/m2/day day every 3 weeks CLCR>60ml/min mg/m2/day CLCR39~60ml/min 1.5 mg/m2/day (未接受过强烈化疗) 1.0 mg/m2/day (接受过强烈化疗) CLCR 20~39ml/min 1.0 mg/m2/day (未接受过强烈化疗) 0.75 mg/m2/day(接受过强烈化疗)
 1.0 mg/m2/day (接受过强烈化疗) CLCR 20~39ml/min. 1.0 mg/m2/day (未接受过强烈化疗) 0.75 mg/m2/day(接受过强烈化疗)")
96
铂类化合物

97
分布:肝、肾、卵巢、子宫、皮肤、骨。90%药物 与蛋白结合→共价键结合(不可逆) 白蛋白减少→剂量减少
顺 铂 给药方式:静脉、动脉、腔内给药 分布:肝、肾、卵巢、子宫、皮肤、骨。90%药物 与蛋白结合→共价键结合(不可逆) 白蛋白减少→剂量减少 肾脏消除(20~70% )24h %-34% d %-45% CLCR60ml/min % CLCR45ml/min %
 白蛋白减少→剂量减少. 肾脏消除(20~70% )24h 19%-34% 5d 25%-45% CLCR60ml/min 75% CLCR45ml/min 50%")
98
毒性 肾功能:剂量相关,并且有聚集性, Cfree 大小。 大剂量:水化疗法 利尿剂 高渗3%,防止肾小管摄取顺铂 胃肠道 骨髓抑制:剂量相关>50mg/m d-23d 4W-6W恢复 可逆性 神经毒性:>300mg/m2 ,多见

99
药物相互作用:顺铂增加博莱霉素的毒性; 吩噻嗪类药物顺铂耳毒性,与顺铂合用要注意

100
卡铂 药代动力学特点:77%经肾脏排泄,CL主要由肾小球滤过率决定,AUC与CL有关而与BSA无关。 卡铂根据AUC个体化给药的公式
Calvert and co-workers 公式 Dose=AUC CL Dose=AUC free (GFR+25)
")
101
CLCR-CG=1.23(140-age) BW (0.85 if female) SCR *Chatelut 公式:
*51Cr-EDTA法 繁琐,但准确性高 *收集24h尿量法 不准确 *Cockcroft-Gault 公式: CLCR-CG=1.23(140-age) BW (0.85 if female) SCR *Chatelut 公式: Clcarboplatin=0.134+ 218 BW ( AGE) ( gender)
 BW (0.85 if female) SCR. *Chatelut 公式: Clcarboplatin= BW ( AGE) ( gender)")
102
欧洲研究表明,应用Calvert公式结合CLCR-CG和CLCR-24计算的AUC分别有10%高误差和10%的低误差,认为Chatelut公式较能准确的预测AUC 。
日本研究刚好相好,认为Calvert和CLCR-CG公式较能准确的预测AUC。 中国研究Calvert和CLCR-CG公式较能准确的预测AUC,能达到个体化给药的目的。

103
奥沙利铂 40%药物→红细胞结合,红细胞降低,调整剂量 30~50%→肾脏排泄,CLCR(27~57ml/min)不需调整剂量
研究表明CLCR减少,延长t1/2,但并不增加毒性。 与5-FU合用,药代动力学不改变。

104
大分子抗体 小分子化合物 靶向治疗抗肿瘤药 曲妥珠单抗 吉非替尼 西妥昔 甲磺酸伊马替尼 贝伐单抗 埃罗替尼 利妥昔单抗 万珂
大分子抗体 小分子化合物 曲妥珠单抗 吉非替尼 西妥昔 甲磺酸伊马替尼 贝伐单抗 埃罗替尼 利妥昔单抗 万珂 Alemtuzumab 吉妥单抗

105
西妥昔(C225) ◆机制:与人表皮生长因子受体(EGFR)胞外区特异性结合,抑制与受体相关的激酶的磷酸化和活化,从而抑制细胞生长、诱导调亡、减少基质金属蛋白酶和血管内皮因子的产生。 ◆药代动力学特点: T1/2 :41~213 h 分布容积与剂量无关,为2~3 L/m2 当剂量从20mg/ m2增至200mg/ 时,清除率由0.08L/( m2.h)降至0.02L/( m2.h ) 经蛋白代谢途径降解
降至0.02L/( m2.h ) 经蛋白代谢途径降解.")
106
吉非替尼 (Gifitinib) 埃罗替尼(Erlotinib)
◆机制:竞争性结合EGFR胞内区酪氨酸激酶催化区的mg-ATP结合点,抑制酪氨酸激酶活性,阻断激酶及底物的磷酸化,切断细胞生存和异常增殖及新生血管形成,促其调亡。 埃罗替尼(Erlotinib) ◆机制:通过在细胞内与三磷酸腺苷竞争结合受体酪氨酸激酶的胞内催化部位,抑制磷酸化反应,从而阻滞向下游增殖信号传导,抑制肿瘤细胞配体依赖或配体外依赖的HER-1/EG-FR的活性,达到抑制癌细胞增殖作用
 ◆机制:通过在细胞内与三磷酸腺苷竞争结合受体酪氨酸激酶的胞内催化部位,抑制磷酸化反应,从而阻滞向下游增殖信号传导,抑制肿瘤细胞配体依赖或配体外依赖的HER-1/EG-FR的活性,达到抑制癌细胞增殖作用.")
107
◆药代动力学特点 吉非替尼 埃罗替尼 口服吸收较慢,7~10 d达Css 7d 食物不影响其吸收 F :57%
吉非替尼 埃罗替尼 口服吸收较慢,7~10 d达Css d 食物不影响其吸收 F :57% T1/2为41 h 蛋白结合率:92% Cmax/Cmin 2~ 代谢产物具有活性 血浆总清除率500 ml/min 组织内分布广泛 吸烟不影响其代谢 粪便排泄

108
Effect of smoking on imatinib pharmacokinetics
Smokers nonsmokers CL ± ±4.6 V ± ±116.9 AUC ± ±116.9 T1/ ± ±6.0

109
Pharmacokinetic of gefitinib predicts the antitumor activity for advanced non-small cell lung cancer
high D8/D low D8/D hazard rate TTP d d never smoking smoking hazard rate TTP d d 腺癌 非腺癌 TTP d d

110
抗癌药物根据PK参数给药 抗癌药物常规给药是依据体表面积(BSA)给药 依据:BSA与基本代谢速率相关 BSA与血容量成正比
有些药物如sulfadiazine依据BSA 给药,血药浓度与剂量成正比。

111
不足之处: Gehan和George 应用DuBois公式计算401例人的BSA,直接测定BSA ,发现DuBois公式有15%过高偏差。
BSA与PK参数无关 如果按BSA给药,不同病人的Cl和AUC应该相同,但实际上AUC和CL存在很大的变异。

112
PK参数与毒性的关系 5-FU AUC>30mg.h/L associated with gastrointestinal
toxicity and leukopenia Css>1.5umol/L or low CL associated leukopenia Carboplatin AUC of platinum correlated with throbocytopenia Doxorubicin doxorubicin AUC correlated with leukopenis, and thrombocytopenia CPT Diarrhea correlated with AUC of CPT-11 and SN-38 Topotecan AUC and Css correlated with hematologic toxicity AUC correlated with myelotoxicity and severity of diarrhea Methotrexate Mucositis and myleosuppression correlated with 48-hour methotrexate level >0.9umol/L

113
PK参数与疗效的关系 Methotrexate High CL associated with increased relapse rate
Doxorubicin hour plasma level after bolus correlated with death during induction and length of remission Ara-c Ara-CTP retention in leukemic lbasts at 4-hour correlated with rate and duration of complete response Etoposide Response=5/11 for pateints with Css of >2ug/ml, 1/10 for 1ug/ml (14-day infusion) No correlation between drug level and response (oral) 5-FU and folinic acid Patients failing to respond had lower plasma and folate concentration v those who respond 5-FU Gowth in liver metastases associted with low AUC
 No correlation between drug level and response (oral) 5-FU and folinic acid Patients failing to respond had lower. plasma and folate concentration v those who respond. 5-FU Gowth in liver metastases associted with low AUC.")
114
PK参数与抗癌药物的关系 PK参数预测毒性优于BSA PK参数预测毒性优于预测疗效 预测疗效仅限于对化疗敏感的肿瘤如淋巴瘤、急淋

115
定义:综合了药代动力学原理,新的分析技术使监测药物在尽可能短的时间内达到最理想的药理效应,使药物的毒性作用最小。
治疗药物监测(TDM) 定义:综合了药代动力学原理,新的分析技术使监测药物在尽可能短的时间内达到最理想的药理效应,使药物的毒性作用最小。 抗癌药物进行TDM的必要性: 1、治疗指数窄、毒性反应强 2、肿瘤患者的药代动力学差异大 3、肿瘤患者肝、肾功能化疗前后变化较大
 定义:综合了药代动力学原理,新的分析技术使监测药物在尽可能短的时间内达到最理想的药理效应,使药物的毒性作用最小。 抗癌药物进行TDM的必要性: 1、治疗指数窄、毒性反应强. 2、肿瘤患者的药代动力学差异大. 3、肿瘤患者肝、肾功能化疗前后变化较大.")
116
谢 谢

Similar presentations



>")








>")







