Download presentation
1
4.7 维生素的测定 概述 脂溶性维生素A、E的测定 水溶性维生素 B、C的测定

2
4.7.1 概述 (1)维生素的分类及生理功能 (2)测定的目的和意义 (3)维生素的测定方法
维生素的分类及生理功能 (2)测定的目的和意义 (3)维生素的测定方法")
3
(1)维生素的分类及生理功能 功能: 维生素是维持人体正常生命活动所必需的一类小分子有机化合物,结构复杂,种类多。目前已经确认的有30多种,其中有20种被认为与维持人体健康和促进生长发育是至关重要的。 主要是通过作为辅酶的成分调节代谢过程,人体对它的需要量极少,但作用非常大。 一般在体内不能合成或合成量不能满足生理需要,必须经常从食物中摄取;长期缺乏任何一种维生素都会导致相关新陈代谢的紊乱,出现各种特有的症状。

4
分类: 按溶解性能可将它们分成两大类: 一类是能溶在脂肪或脂溶性溶剂中的,叫脂溶性维生素(如A、D、E、K等);
另一类是能溶解在水中的,叫水溶性维生素(如B1、B2、B6、C、 B12等)。
。")
5
(2)测定的目的和意义 可评价食品的营养价值; 开发利用富含维生素的食品;
多数维生素的性质不稳定,常常作为强化剂在食品工业的某些产品中使用。可以研究食品在不同的加工、储存条件下的稳定性,进而指导人们制定合理的工艺条件,减少维生素的损失; 起到监督维生素强化食品的剂量,以防摄入过多的维生素而引起中毒。

6
微生物法:基于某种微生物生长需要特定的维生素,方法特异性强、灵敏度高、不需要特殊仪器,样品不需经特殊处理,但只能测定水溶性维生素。
(3)维生素的测定方法 微生物法:基于某种微生物生长需要特定的维生素,方法特异性强、灵敏度高、不需要特殊仪器,样品不需经特殊处理,但只能测定水溶性维生素。 化学法:比色法、滴定法 仪器法:色谱法、荧光法、酶法、免疫法等。特别是HPLC可用于大多数维生素的测定,并且在某些条件下可同时分析几种维生素,但分析费用较高。 应根据不同的食品基质和条件选择不同的分析方法
维生素的测定方法. 微生物法:基于某种微生物生长需要特定的维生素,方法特异性强、灵敏度高、不需要特殊仪器,样品不需经特殊处理,但只能测定水溶性维生素。 化学法:比色法、滴定法. 仪器法:色谱法、荧光法、酶法、免疫法等。特别是HPLC可用于大多数维生素的测定,并且在某些条件下可同时分析几种维生素,但分析费用较高。 应根据不同的食品基质和条件选择不同的分析方法.")
7
重点讲解的方法: 高效液相色谱法(HPLC法)同时测定脂溶性维生素A和维生素E 比色法测定维生素A HPLC法测定胡萝卜素 滴定法和比色法测定维生素C 荧光法测定 B1、B2
同时测定脂溶性维生素A和维生素E 比色法测定维生素A HPLC法测定胡萝卜素 滴定法和比色法测定维生素C 荧光法测定 B1、B2")
8
4.7.2 维生素A、E的测定—HPLC法 (GB/T 5009.82)
VA的性质: 维生素A是β-紫罗酮环与一元醇所组成的一类化合物及其衍生物的总称。通常所说的维生素A是指视黄醇或视黄醇醋酸酯。 ⑴ 因有许多不饱和链,故见光易分解; ⑵ 在缺氧情况下,对热较稳定,对光特别敏感。测定速度要快。 ⑶ 不溶于水,溶于有机溶剂,对碱稳定

9
VE的性质:又称生育酚,目前已经确认 的有8种异构体,最常见的有α、β、γ、δ-生育酚。溶于脂溶性溶剂,对热稳定,酸性环境中比碱性环境中稳定,无氧条件下,对热、光及碱性环境中也相对稳定。VE广泛分布在各种粮食的胚和植物油中。

10
测定原理: (1)液相色谱的原理:液相色谱是采用液体为流动相的一种色谱法。高效液相色谱仪由高压泵、进样装置、色谱柱、检测器、记录和数据处理装置等部分组成。高压泵以恒定的流量,从贮液容器吸取作为流动相的溶剂输送到色谱柱,试样由进样装置导入,随流动相在色谱柱内进行分离。分离后的组分进入检测器检测,并用记录仪绘出色谱图,或与其他数据处理装置相连,由数据处理系统进行数据处理 色谱图:某一波长下各个不同时刻的吸光度值描点作图。
液相色谱的原理:液相色谱是采用液体为流动相的一种色谱法。高效液相色谱仪由高压泵、进样装置、色谱柱、检测器、记录和数据处理装置等部分组成。高压泵以恒定的流量,从贮液容器吸取作为流动相的溶剂输送到色谱柱,试样由进样装置导入,随流动相在色谱柱内进行分离。分离后的组分进入检测器检测,并用记录仪绘出色谱图,或与其他数据处理装置相连,由数据处理系统进行数据处理. 色谱图:某一波长下各个不同时刻的吸光度值描点作图。")
11
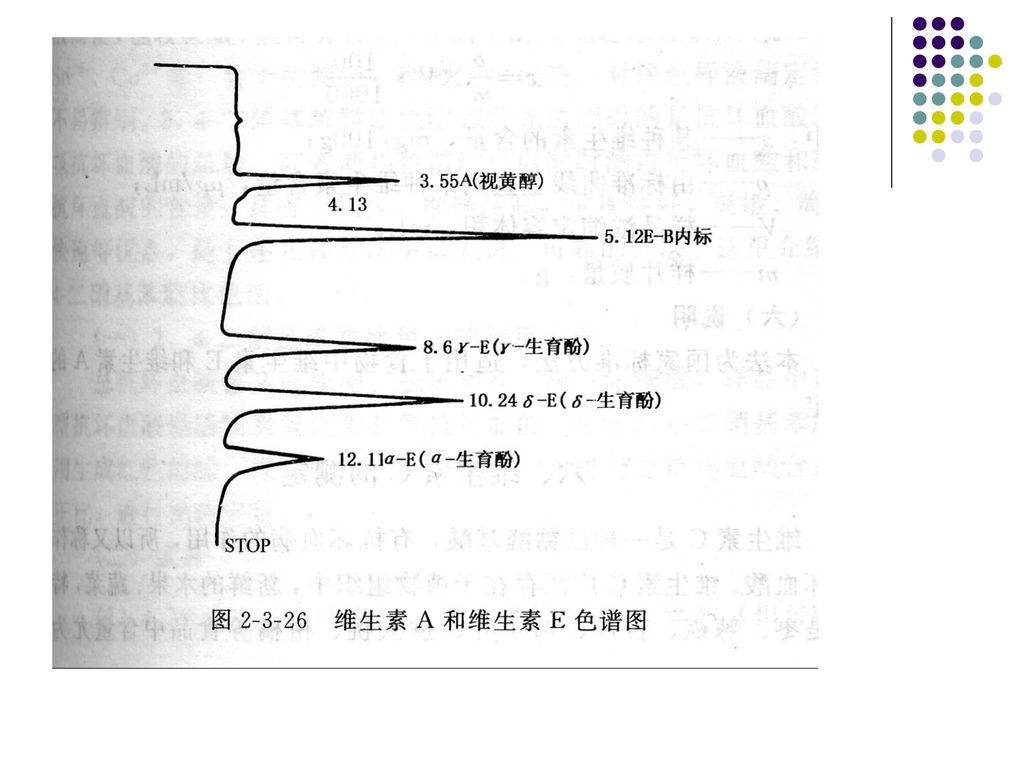
(2)高效液相色谱检测样品中的维生素A、E的原理: 样品中的维生素E及维生素A经皂化提取处理后,将其从不皂化部分提取至有机溶剂中。用高效液相色谱法C18反相柱将维生E和维生素A分离,经紫外检测器检测,并用内标法定量
高效液相色谱检测样品中的维生素A、E的原理: 样品中的维生素E及维生素A经皂化提取处理后,将其从不皂化部分提取至有机溶剂中。用高效液相色谱法C18反相柱将维生E和维生素A分离,经紫外检测器检测,并用内标法定量")
12
(3)什么是内标法? 取标准被测成分,按依次增加或减少的已知阶段量,各自分别加入各单体所规定的定量内标准物质中,调制成标准溶液。分别取此标准液的一定量注入色谱柱,根据色谱图取标准被测成分的峰面积或峰高和内标物质的峰面积或峰高的比例为纵坐标,取标准被测成分量和内标物质量之比或标准被测成分量为横坐标,制成标准曲线。 然后按单体中所规定的方法调制试样液,调制试样液时,预先加入与调制标准溶液等量的内标物质,然后按制作标准曲线时的同样条件下得出色谱图,求出被测成分的峰面积或峰高和内标物质的峰面积或峰高之比,再按标准曲线来求出被测成分的含量。

13
内标物的选取原则:能与被测成分完全分离,但其保留时间又尽可能接近被测成分的稳定的物质。

14
试剂 (1)无水乙醚:不含有过氧化物。 (2)无水乙醇:不得含有醛类物质。 (3)无水硫酸钠。 (4)甲醇:重蒸后使用。
(5)重蒸水:水中加少量高锰酸钾,临用前蒸馏。 (6)抗坏血酸溶液(100g/L):临用前进行配制。 (7)氢氧化钾溶液(50g/100g):取50g氢氧化钾,溶于50g水中,混匀
重蒸水:水中加少量高锰酸钾,临用前蒸馏。 (6)抗坏血酸溶液(100g/L):临用前进行配制。 (7)氢氧化钾溶液(50g/100g):取50g氢氧化钾,溶于50g水中,混匀.")
15
标准溶液的配制 (1)维生素 A标准液:视黄醇(纯度85%) 或视黄醇乙酸酯(纯度90%)经皂化处理后使用。 用脱醛乙醇溶解维生素A标准品,使其浓度大约为lmg/mL。临用前用紫外分光光度法标定其准确浓度。 (2)维生素E标准液:a-生育酚(纯度95%),γ-生育酚(纯纯95%), δ-生育酚(纯度95%)。用脱醛乙醇分别溶解以上三种维生素E标准品,使其浓度大约为lmg/mL,临用前用紫外分光光度法分别标定此三种维生素E的准确浓度。 (3)内标溶液:称取苯并芘(纯度98%),用脱醛乙醇配制成10ug/mL 的内标溶液。
维生素E标准液:a-生育酚(纯度95%),γ-生育酚(纯纯95%), δ-生育酚(纯度95%)。用脱醛乙醇分别溶解以上三种维生素E标准品,使其浓度大约为lmg/mL,临用前用紫外分光光度法分别标定此三种维生素E的准确浓度。 (3)内标溶液:称取苯并芘(纯度98%),用脱醛乙醇配制成10ug/mL 的内标溶液。")
16
仪器和设备 (1)高效液相色谱仪带紫外分光检测器。 (2)旋转蒸发器。 (3)高速离心机(带小塑料离心管) (4)高纯氮气。
(5)紫外分光光度计。
紫外分光光度计。")
17
样品处理: 皂化→提取→洗涤→浓缩→ 定容

18
(1)皂化: 称取适量样品于三角瓶中,加30mL无水乙醇,振摇使样品分散。加入 5mL100g/L抗坏血酸溶液和 2.0mL苯并芘内标液,混匀,加 10mL氢氧化钾溶液,沸水回流30min,使皂化完全,皂化后立即放入冰水中冷却。 思考:皂化时加入Vc的作用是什么?

19
(2)提取: 将皂化后的样品移入分液漏斗中,用50mL水分2-3次洗皂化瓶,洗液并入分液漏斗中。用100mL无水乙醚分2次洗皂化瓶及残渣,乙醚液并入分液漏斗中。轻轻振摇2min,静置分层,弃去水层。
提取: 将皂化后的样品移入分液漏斗中,用50mL水分2-3次洗皂化瓶,洗液并入分液漏斗中。用100mL无水乙醚分2次洗皂化瓶及残渣,乙醚液并入分液漏斗中。轻轻振摇2min,静置分层,弃去水层。")
20
(3) 洗涤: 每次用约50mL水将乙醚液洗至中性,需洗4~5次
 洗涤: 每次用约50mL水将乙醚液洗至中性,需洗4~5次")
21
(4)浓缩: 将乙醚提取液经无水硫酸钠(约5g)滤入150 mL旋转蒸发瓶内,用约15mL乙醚冲洗分液漏斗及无水硫酸钠2次,并入蒸发瓶内,于55℃水浴中减压蒸馏并回收乙醚,醚剩下约2mL时,取下蒸发瓶。
浓缩: 将乙醚提取液经无水硫酸钠(约5g)滤入150 mL旋转蒸发瓶内,用约15mL乙醚冲洗分液漏斗及无水硫酸钠2次,并入蒸发瓶内,于55℃水浴中减压蒸馏并回收乙醚,醚剩下约2mL时,取下蒸发瓶。")
22
(5)定容: 用氮气吹干乙醚,随即加入2mL乙醇,充分混合,溶解提取物。5000r/min离心5min,上清液供色谱分析用。
定容: 用氮气吹干乙醚,随即加入2mL乙醇,充分混合,溶解提取物。5000r/min离心5min,上清液供色谱分析用。")
23
液相色谱推荐条件: 分析柱:C18反相柱 5µm,4.6mm×25Cm; 流动相:甲醇:水=98:2,混匀,临用前脱气;
紫外检测器波长:300nm; 进样量:20 µ L; 流速:1.70mL/min。

24
标准曲线的制备 (1)维生素A和维生素E标准溶液的标定:
比吸光系数E (1%,cm) 测定波长(nm) 视黄醇 1835 325 γ-生育酚 71 294 δ-生育酚 92.8 298 a-生育酚 91.2
 测定波长(nm) 视黄醇 γ-生育酚 δ-生育酚 a-生育酚")
25
标准溶液浓度计算: p---某维生素浓度,mg/mL; A---维生素的平均紫外吸光值; E---某种维生素l%比吸光系数;
F---稀释倍数。

26
(2)标准曲线的制备: 本方法采用内标法定量。把高、低两个浓度的
维生素A、γ-生育酚、a-生育酚、 δ-生育酚及内标苯并芘液混合,(其内标苯并芘的浓度值相同)将二种浓度的混合标准进行色谱分析,结果见色谱图。以维生素峰面积与内标物峰面积之比为纵坐标,维生素的浓度(ug/mL)为横坐标绘制标准曲线。
将二种浓度的混合标准进行色谱分析,结果见色谱图。以维生素峰面积与内标物峰面积之比为纵坐标,维生素的浓度(ug/mL)为横坐标绘制标准曲线。")
28
样品分析: 取样品处理液20 µ l,用标准溶液同样的条件进行色谱分析,绘制色谱图。 定性:根据保留时间定性
定量:根据色谱图求出某种维生素的峰面积与内标物峰面积的比值,以此比值在标准曲线上查得待测维生素的含量。

29
计算: 式中: X---某种维生素的含量,mg/100g; P---由标准曲线上查到的某种维生素含量, µg/mL;
V---样品浓缩后定容体积,ml; m--样品质量,g。

30
说明及讨论 (1)维生素极易被光破坏,实验操作应在微弱光线下进行,或使用棕色玻璃仪器。
(2)乙醚为溶剂的萃取体系,易发生乳化现象。在提取、洗涤操作中,不要用力过猛,若发生乳化,可加几滴乙醇破乳。 (3)本法是国家标准检验方法,适用于各种食物和饲料中维生素A和维生素E的同时测定。 (4)本法不能将β-生育酚γ-生育酚分开,所以γ-生育酚的色谱峰中含有β-生育酚。
乙醚为溶剂的萃取体系,易发生乳化现象。在提取、洗涤操作中,不要用力过猛,若发生乳化,可加几滴乙醇破乳。 (3)本法是国家标准检验方法,适用于各种食物和饲料中维生素A和维生素E的同时测定。 (4)本法不能将β-生育酚γ-生育酚分开,所以γ-生育酚的色谱峰中含有β-生育酚。")
31
IU(国际单位)与质量的换算 1IUVa=0.33μg 1IUβ-胡萝卜素=0.6μg 1Ug Vd=40IU 1IUVd=0.025μg
1IU维生素D=0.025ug维生素D3 1IU Ve=1mg Ve(DL-a-生育酚醋酸酯) 1mgDL-a-生育酚=1.1IU Ve 1mgD-a-生育酚=1.49 IU Ve 1mgD-a-生育酚醋酸酯=1.36 IU Ve
 1mgDL-a-生育酚=1.1IU Ve. 1mgD-a-生育酚=1.49 IU Ve. 1mgD-a-生育酚醋酸酯=1.36 IU Ve.")
32
4.7.3 维生素A的测定(三氯化锑比色法) 原理: VA+三氯化锑→蓝色物质,于620nm测吸光度,与标准比较定量。 试剂:
试剂: 无水Na2SO4、乙酸酐:吸水 乙醚:抽提 乙醇、KOH:皂化反应 三氯甲烷:溶剂 三氯化锑-三氯甲烷:反应试剂 酚酞:用于鉴别洗涤液中有无碱

33
步骤: (1)预处理:皂化、提取、洗涤、浓缩 (2)制备标准曲线:用VA标准液绘制,用CHCl3调零。
注意在移入光路前,迅速加入三氯化锑,6秒内测定 (3)样品测定:用样品溶液代替标准液溶液。
样品测定:用样品溶液代替标准液溶液。")
34
说明 (1) 萃取时,不要用力过猛,避免发生乳化。 (2)氯仿中必须无水,因三氯化锑遇水会出现沉淀,干扰比色测定。
(3)由于三氯化锑与VA所产生的兰色物质不稳定,注意在移入光路前,迅速加入三氯化锑,6秒内测定。 (4)三氯化锑腐蚀性强,且遇水生成白色沉淀,实验用过的仪器要先用稀盐酸浸泡后再清洗。
由于三氯化锑与VA所产生的兰色物质不稳定,注意在移入光路前,迅速加入三氯化锑,6秒内测定。 (4)三氯化锑腐蚀性强,且遇水生成白色沉淀,实验用过的仪器要先用稀盐酸浸泡后再清洗。")
35
植物油中VE的测定(文献方法) 样品处理:准确称取1g左右的样品用25mL丙酮溶解。 仪器条件:流动相:乙腈+甲醇=80+20
流速:1.2ml/min 色谱柱: C18反相柱 波长:298nm 此法可以同时测定 γ-生育酚、a-生育酚、 δ-生育酚

36
4.7.4 β-胡萝卜素的测定(HPLC法) 试样中的β-胡萝卜素,用石油醚+丙酮混合液提取,经三氧化铝柱纯化,然后以高效液相色谱法测定,以保留时间定性,以峰高或峰面积定量。 注意:浓缩提取液时,防止蒸干,避免胡萝卜素在空气中氧化或因高温、紫外线直射等破坏。
 试样中的β-胡萝卜素,用石油醚+丙酮混合液提取,经三氧化铝柱纯化,然后以高效液相色谱法测定,以保留时间定性,以峰高或峰面积定量。 注意:浓缩提取液时,防止蒸干,避免胡萝卜素在空气中氧化或因高温、紫外线直射等破坏。")
37
维生素B1的测定—荧光法(GB/T ) 原理:硫胺素(VB1)在碱性铁氰化钾溶液中被氧化为嘧噻色素,在紫外线照射下,嘧噻色素发出荧光。在给定条件下,以及没有其他荧光物质干扰时,荧光强度与嘧噻色素成正比,即与溶液中硫胺素量成正比。 试样在硫酸作用下,加热提取VB1,冷却后用淀粉酶处理使VB1分离,然后以所得溶液做测定。 测定步骤:提取(水解、酶解)→净化→氧化→测定 水解、酶解的作用:使结合态VB1变为游离态VB1
→净化→氧化→测定. 水解、酶解的作用:使结合态VB1变为游离态VB1.")
38
维生素B1的测定—HPLC法 维生素B1通常采用反相键合相色谱法进行分离,利用紫外检测器或荧光检测器进行测定。利用荧光检测器检测时,应首先使从样品中提取的维生素B1氧化成硫色素,然后转入正丁醇或异丁醇中,再进行HPLC分析。

39
4.7.6 维生素B2的测定——荧光法 原理:试样在稀盐酸中水解、酶解,调整 PH值,过滤除去蛋白质等物质;试样溶液经高锰酸钾氧化,除去干扰物,再经硅镁吸附剂的柱层析,提纯的核黄素在波长525nm下发生黄绿色荧光,在稀溶液中其荧光强度与核黄素的浓度成正比。再加入低亚硫酸钠,将VB2还原成无荧光的物质,然后再测定试液中残余荧光杂质的荧光,两者相差即为食品中核黄素所产生的荧光强度。 测定核黄素的方法还有:微生物法和HPLC法。

41
4.7.7 维生素C(抗坏血酸)的测定 维生素C的生理功能及性质 维生素C是一种己糖醛基酸,有抗坏血病的作用,所以又称作抗坏血酸。
在食品中,这三种形式均有存在,但主要是前两者,故许多国家的食品成分表均以抗坏血酸和脱氢抗坏血酸的总量表示。

42
还原型维生素 C有强还原性,有抑制多酚氧化酶作用,故常添加于啤酒、果汁等食品中作抗氧化剂。
在新鲜的水果蔬菜,特别是枣、辣椒、苦瓜、猕猴桃、柑橘等食品中含量尤为丰富。

43
维生素C 的测定方法 2,6—二氯靛酚滴定法 2,4—二硝基苯肼比色法 荧光法 高效液相色谱法

44
(1)2,6—二氯靛酚滴定法 测定的是还原型抗坏血酸的含量 该方法简便、灵敏, 但特异性差,样品中的其他还原性物质(如铁离子、铜离子等)会干扰测定,使结果偏高,对色深样液的滴定终点不易辨别。
2,6—二氯靛酚滴定法 测定的是还原型抗坏血酸的含量 该方法简便、灵敏, 但特异性差,样品中的其他还原性物质(如铁离子、铜离子等)会干扰测定,使结果偏高,对色深样液的滴定终点不易辨别。")
45
(2)2,4—二硝基苯肼比色法和荧光法 测得的是抗坏血酸和脱氢抗坏血酸的总量。 苯肼法操作复杂,特异性差,易受共存物质的影响; 荧光法受干扰的影响较小,结果较准确,重现性好,但操作复杂。
2,4—二硝基苯肼比色法和荧光法 测得的是抗坏血酸和脱氢抗坏血酸的总量。 苯肼法操作复杂,特异性差,易受共存物质的影响; 荧光法受干扰的影响较小,结果较准确,重现性好,但操作复杂。")
46
(3)高效液相色谱法 可以同时测得抗坏血酸和脱氢抗坏血酸的含量。 具有干扰少,准确度高,重现性好,灵敏、简便、快速等优点,是上述几种方法中最先进、可靠的方法。但分析成本较高。 主要介绍维生素C常用的测定方法2,6—二氯靛酚滴定法、苯肼比色法
高效液相色谱法 可以同时测得抗坏血酸和脱氢抗坏血酸的含量。 具有干扰少,准确度高,重现性好,灵敏、简便、快速等优点,是上述几种方法中最先进、可靠的方法。但分析成本较高。 主要介绍维生素C常用的测定方法2,6—二氯靛酚滴定法、苯肼比色法.")
47
(一)2,6-二氯靛酚滴定法 以新鲜果蔬为样品进行讲解和演示 原理:
还原型抗坏血酸可以还原染料2,6—二氯靛酚。该染料在酸性溶液中是粉红色(在中性或碱性溶液中呈蓝色),被还原后颜色消失;还原型抗坏血酸还原染料后,本身被氧化成脱氢抗坏血酸。在没有杂质干扰时,一定量的样品提取液还原标准染料液的量,与样品中抗坏血酸含量成正比。
,被还原后颜色消失;还原型抗坏血酸还原染料后,本身被氧化成脱氢抗坏血酸。在没有杂质干扰时,一定量的样品提取液还原标准染料液的量,与样品中抗坏血酸含量成正比。")
48
试剂 ① 1%草酸溶液 ② 2%草酸溶液 ③ 抗坏血酸标准溶液 ④ 2,6—二氯靛酚溶液
⑤ 0.001mol/L碘酸钾标准溶液 :精确称取干燥的碘酸钾0.3567g,用水稀释至100ml,取出1ml,用水稀至100ml, 此溶液1ml相当于抗坏血酸0.088mg。 ⑥ 1%淀粉溶液 ⑦ 6%碘化钾溶液

49
抗坏血酸标准溶液的配制及标定: 配制:准确称取20mg抗坏血酸,溶于1%的草酸中,并稀释至100ml,置冰箱中保存。用时取出5ml,置于50ml容量瓶中,用1%草酸溶液定容,配成0.02mg/ml的抗坏血酸标准溶液。 标定:吸取标准使用液 5ml于三角瓶中,加入 6%碘化钾溶液0.5ml、1%淀粉溶液3滴,以0.00lmol/L碘酸钾标准溶液滴定,终点为淡蓝色。

50
C---抗坏血酸标准溶液的浓度,mg/mL;
V1---滴定时消耗 0.001mol/L碘酸钾标准溶液的体积,mL; V2---滴定时所取抗坏血酸的体积,mL; lmL0.001mol碘酸钾标准溶液相当于抗坏血酸的量,mg/mL

51
2,6-二氯靛酚溶液的配制和标定 配制:称取2,6—二氯靛酚50mg,溶于200mL含有52mg碳酸氢钠的热水中,待冷,置于冰箱中过夜。次日过滤于250mL棕色容量瓶中,定容,在冰箱中保存。每星期标定1次。 标定:取5ml已知浓度的抗坏血酸标准溶液,加入1%草酸溶液5ml,摇匀,用2,6—二氯靛酚溶液滴定至溶液呈粉红色,在15s不褪色为终点。

52
计算: 式中: T---每毫升染料溶液相当于抗坏血酸的毫克数,mg/mL; C---抗坏血酸的浓度,mg/mL;
V1---抗坏血酸标准溶液的体积,ml; V2---消耗 2,6- 二氯靛酚的体积,ml

53
测定步骤 ① 提取: 鲜样的制备: 称100g鲜样,加等量的2%草酸溶液,于组织捣碎机中打成匀浆。取10~40g匀浆于100ml 容量瓶内,用1%草酸稀释至刻度,混合均匀。 干样的制备: 称1~4g干样放入乳钵内加 l%草酸溶液磨成匀浆,倒入100ml容量瓶中,用 1%草酸稀释至刻度。过滤上述样液,不易过滤的可用离心机沉淀后,倾出上清液过滤备用。

54
②滴定: 吸取 5~10ml滤液,快速用 2,6-二氯靛酚溶液滴定,直到红色不能立即消失,而后再尽快地一滴一滴的加入(样品中可能存在其他还原性杂质,但一般杂质还原染料的速度均比抗坏血酸慢),以呈现的粉红色在15s内不消失为终点。同时做空白。
,以呈现的粉红色在15s内不消失为终点。同时做空白。")
55
③结果计算 T---1mL染料溶液相当于抗坏血酸标准溶液的量,mg/mL; V--滴定样液时消耗染料的体积,mL;
式中:x---样品中抗坏血酸含量,mg/100g; T---1mL染料溶液相当于抗坏血酸标准溶液的量,mg/mL; V--滴定样液时消耗染料的体积,mL; V0---滴定空白时消耗染料的体积,mL; m---滴定时所取滤液中样品的质量,g。

56
说明 ① 所有试剂最好用重蒸馏水配制。 ② 样品采取后,应浸泡在已知量的2%草酸溶液中,以防止维生素C氧化损失。操作过程要迅速,防止抗坏血酸被氧化。 ③ 若测动物性样品,须用10%三氯乙酸代替2%草酸溶液提取。 ④ 若样品滤液颜色较深,影响滴定终点观察,可加入白陶土再过滤,过滤后要迅速滴定。 ⑤ 若样品中含有Fe2+、Cu2+、Sn2+、亚硫酸盐、硫代硫酸盐等还原性杂质时,会使结果偏高。

57
(二) 2,4二硝基苯肼比色法 原理 总抗坏血酸包括还原型、脱氢型和二酮古乐糖酸,样品中还原型抗血酸经活性炭氧化为脱氢抗坏血酸,再与2,4-二硝基苯肼生成红色的脎,在浓硫酸的脱水作用下,可转变为桔红色的无水化合物—双-2,4-二硝基苯肼,在浓硫酸中显色稳定,吸光度的大小与总抗坏血酸含量成比,进行比色定量。

58
操作过程: 样品处理→氧化→ 呈色→ 硫酸处理 → 比色→ 数据处理 (1)样品制备:同2,6-二氯靛酚滴定法
(2)氧化处理:取 25mL上述滤液,加入 2g活性炭,振摇 lmin,过滤,弃去最初数毫升滤液。取10mL此氧化提取液,加入 10mL 20g/L的硫脲溶液,混匀。
氧化处理:取 25mL上述滤液,加入 2g活性炭,振摇 lmin,过滤,弃去最初数毫升滤液。取10mL此氧化提取液,加入 10mL 20g/L的硫脲溶液,混匀。")
59
于三个试管中各加入4mL经氧化的样品稀释液,一个试管作为空白,在其余试管中加入1.0mL
(3)呈色反应: 于三个试管中各加入4mL经氧化的样品稀释液,一个试管作为空白,在其余试管中加入1.0mL 20g/L 2,4-二硝基苯肼溶液,将所有试管放入(37士0.5)℃恒温箱或水浴中,保温3h。3h后取出,除空白管外,将所有试管放入冰水中。空白管冷到室温,然后加入 1.0mL 20g/L 2,4-二硝基苯肼溶液,在室温中放置10~15min后放入冰水内。其余步骤同样品。
呈色反应: 于三个试管中各加入4mL经氧化的样品稀释液,一个试管作为空白,在其余试管中加入1.0mL. 20g/L 2,4-二硝基苯肼溶液,将所有试管放入(37士0.5)℃恒温箱或水浴中,保温3h。3h后取出,除空白管外,将所有试管放入冰水中。空白管冷到室温,然后加入 1.0mL 20g/L 2,4-二硝基苯肼溶液,在室温中放置10~15min后放入冰水内。其余步骤同样品。")
60
(4)硫酸(9+1)处理 当试管放入冰水后,向每一试管中加入5mL硫酸(9十1)滴加时间至少需要1min,(防止溶液温度升高而使部分有机物分解着色,影响空白值),需边加边摇动试管。将试管自冰水中取出,在室温放置30min后比色。 (5)比色 用1cm比色杯,以空白液调零点,于500nm波长测吸光值。
比色. 用1cm比色杯,以空白液调零点,于500nm波长测吸光值。")
61
①加 2g活性炭于 50ml标准溶液(1mg/mL)中,摇动 1min,过滤。
(6)标准曲线绘制 ①加 2g活性炭于 50ml标准溶液(1mg/mL)中,摇动 1min,过滤。 ②取10mL滤液放入500ml容量瓶中,加 5.0g硫脲,用 10g/L的草酸溶液稀释至刻度,抗坏血酸浓度20μg/mL。 ③取5、10、20、25、40、50、60ml稀释液,分别放入7个100ml容量瓶中,用 10g/L硫脲溶液稀释至刻度,使最后稀释液中抗坏血酸的浓度分别为1、2、4、5、8、10、12 μg /mL。 ④按样品测定步骤进行显色反应并比色。 ⑤以吸光值为纵坐标,以抗坏血酸浓度( μ g/mL)为横坐标绘制标准曲线。
标准曲线绘制. ①加 2g活性炭于 50ml标准溶液(1mg/mL)中,摇动 1min,过滤。 ②取10mL滤液放入500ml容量瓶中,加 5.0g硫脲,用 10g/L的草酸溶液稀释至刻度,抗坏血酸浓度20μg/mL。 ③取5、10、20、25、40、50、60ml稀释液,分别放入7个100ml容量瓶中,用 10g/L硫脲溶液稀释至刻度,使最后稀释液中抗坏血酸的浓度分别为1、2、4、5、8、10、12 μg /mL。 ④按样品测定步骤进行显色反应并比色。 ⑤以吸光值为纵坐标,以抗坏血酸浓度( μ g/mL)为横坐标绘制标准曲线。")
62
计算 式中 X ---总抗坏血酸含量,mg/100g; ρ---由标准曲线查得或由回归方程算得“样品氧化液”中总抗坏血酸的浓度 μg /mL
V---试样用10g/L草酸溶液定容的体积,ml; F---样品氧化处理过程中的稀释倍数; m---试样质量(10g/L草酸溶液定容后的溶液中试样的质量),g。
,g。")
63
说明 (1)本法为国家标准方法,适用于蔬菜、水果及其制品中总抗坏血酸的测定。
(2)活性炭对抗坏血酸的氧化作用,是基于其表面吸附的氧进行界面反应,加入量过低,氧化不充分,测定结果偏低,加入量过高,对抗坏血酸有吸附作用,使结果也偏低。 (3)硫脲可防止抗坏血酸继续氧化,同时促进脎的形成。最后溶液中硫脲的浓度要一致,否则影响测定结果。
活性炭对抗坏血酸的氧化作用,是基于其表面吸附的氧进行界面反应,加入量过低,氧化不充分,测定结果偏低,加入量过高,对抗坏血酸有吸附作用,使结果也偏低。 (3)硫脲可防止抗坏血酸继续氧化,同时促进脎的形成。最后溶液中硫脲的浓度要一致,否则影响测定结果。")
64
(4)试管从冰浴中取出后,因糖类的存在造成显色不稳定,颜色会逐渐加深,30 min后影响将减小,故在加入9+1 的硫酸后30min后准时比色。
(5)测定波长一般在495~540nm,样品杂质多时在540nm 较合适,但灵敏度较最大吸收波长(520nm)下的灵敏度降低30%
测定波长一般在495~540nm,样品杂质多时在540nm 较合适,但灵敏度较最大吸收波长(520nm)下的灵敏度降低30%")
65
HPLC法测定样品中VC含量 优点:选择性强、灵敏度高、快速简单。
样品处理:液体样品可过滤后直接进样;固体样品可用草酸或偏磷酸浸泡或经澄清处理后进样。 推荐色谱条件:氨基柱或反相离子交换柱 流动相:甲醇-水(5+95) 流速:1. 5mL/min 波长:254nm
 流速:1. 5mL/min. 波长:254nm.")
66
作业: 测定维生素A时,为什么要用皂化法处理样品? 维生素A和维生素C的测定中,样品处理和提取有何不同之处?为什么? 继续练习滴定法测定维生素C


、 B 5 (泛酸)、 B 6 、 B 11 (叶酸)、 B 12 (钴胺素)。>")










维生素(维他命):指维持人体正常生命活动所必需的一类天然的有机化合物 。 (二)测定维生素的意义>")







