Download presentation
Presentation is loading. Please wait.

1
第三章 自由基聚合

2
3.1 引言 一、连锁聚合聚合特征: 链引发 链增长 链终止

3
自由基、阳离子和阴离子的产生: 引发剂分解成活性中心时,共价键有两种裂解形式: 均裂和异裂。 均裂的结果产生两个自由基
异裂的结果形成阴离子和阳离子

4
活性中心 ( Reactive Center)
自由基、阴离子和阳离子均有可能作为连锁聚合的活性中心。 自由基-自由基聚合反应 阳离子-阳离子聚合反应 阴离子-阴离子聚合反应 配位(阴离子)聚合
聚合.")
5
链式聚合反应 必须有活性中心,且活性 中心活性较高; 活性中心一旦形成立即以链式反应加上单体单元,快速形成高分子活性链;只有链增长反应才使聚合度增加。 反应体系中总是存在单体、高分子和极少量高分子活性链。

6
一、单体的聚合能力和对不同聚合机理的选择
烯类单体聚合能力的差异和聚合机理的不同主要取决于 双键碳原子上取代基的种类、数量和位置, 取代基的电子效应(诱导效应、共轭效应)和空间位阻效应。
和空间位阻效应。")
7
(一)电子效应 醛、酮中的羰基π键异裂后,具有类似离子的特性,可发生阴离子或阳离子聚合,不能进行自由基聚合。环状单体一般也按阴离子或阳离子机理进行聚合。 烯类单体的碳-碳π键与羰基不同,既可均裂,也可异裂,故可以进行自由基聚合或离子聚合(阴离子聚合、阳离子聚合)。 乙烯基单体取代基的诱导效应和共轭效应能改变双键的电子云密度,对所形成的活性种的稳定性有影响,从而决定着对自由基、阳离子或阴离子聚合的选择性。

8
乙烯基单体(CH2=CHX)的聚合反应性能主要取决于双键上取代基的电子效应。
增大电子云密度,易与阳离子活性种结合 分散正电性,稳定阳离子 因此带给电子基团的烯类单体易进行阳离子聚合,如X = -R,-OR,-SR,-NR2,苯基,乙烯基等。

9
乙烯分子中无取代基,结构对称,因此无诱导效应和共轭效应。只能在高温高压下进行自由基聚合,得到低密度聚乙烯。在配位聚合引发体系引发下也可进行常温低压配位聚合,得到高密度聚乙烯。
丙烯分子上有一个甲基,具有推电子性和超共轭双重效 应,但都较弱,不足以引起阳离子聚合,也不能进行自由基 聚合。只能在配位聚合引发体系引发下进行配位聚合。 其它含有一个烷基的乙烯基单体也具有类似的情况。

10
结论: 1,1取代的异丁烯分子中含有两个甲基,推电子能力大大 增强,可进行阳离子聚合,但不能进行自由基聚合。
含有烷氧基的烷氧基乙烯基醚、苯基的苯乙烯、乙烯基 的丁二烯均可进行阳离子聚合。 结论: 含有1,1-双烷基、烷氧基、苯基和乙烯基的烯烃因推电 子能力较强,可进行阳离子聚合。

11
(2) X为吸电子基团 降低电子云密度,易与富电性活性种结合 分散负电性,稳定活性中心 由于阴离子与自由基都是富电性的活性种,因此带吸电子基团的烯类单体易进行阴离子聚合与自由基聚合,如X = -CN,-COOR,-NO2等; 但取代基吸电子性太强时一般只能进行阴离子聚合。如CH2=C(CN)2
2.")
12
(3) 具有共轭体系的烯类单体 电子云流动性大,易诱导极化,可随进攻试剂性质的不同而取不同的电子云流向,可进行多种机理的聚合反应。因此既可进行自由基聚合,也可进行阴、阳离子聚合。如苯乙烯、 α-苯乙烯、丁二烯、异戊二烯等。
 具有共轭体系的烯类单体 电子云流动性大,易诱导极化,可随进攻试剂性质的不同而取不同的电子云流向,可进行多种机理的聚合反应。因此既可进行自由基聚合,也可进行阴、阳离子聚合。如苯乙烯、 α-苯乙烯、丁二烯、异戊二烯等。")
13
卤素原子既有诱导效应(吸电子),又有共轭效应(推
电子),但两者均较弱,因此既不能进行阴离子聚合,也不 能进行阳离子聚合,只能进行自由基聚合。如氯乙烯、氟乙 烯、四氟乙烯均只能按自由基聚合机理进行。 除了少数含有很强吸电子基团的单体(如偏二腈乙烯、 硝基乙烯)只能进行阴离子聚合外,大部分含吸电子基团的 单体均可进行自由基聚合。
,但两者均较弱,因此既不能进行阴离子聚合,也不. 能进行阳离子聚合,只能进行自由基聚合。如氯乙烯、氟乙. 烯、四氟乙烯均只能按自由基聚合机理进行。 除了少数含有很强吸电子基团的单体(如偏二腈乙烯、 硝基乙烯)只能进行阴离子聚合外,大部分含吸电子基团的. 单体均可进行自由基聚合。")
14
依据单烯CH2=CHX中取代基X电负性次序和聚合倾向的关系排列如下:

15
(二)空间位阻效应 由取代基的体积、数量和位置等因素所引起的空间位阻作用,对单体的聚合能力有显著影响,但不影响其对活性种的选择性。
单取代烯类单体, 即使取代基体积较大,也不妨碍聚合。 1、对于1,1-双取代烯类单体CH2=CXY,1,1双取代的烯类单体,因分子结构对称性更差,极化程度增加,因此更容易聚合。取代基体积较大时例外,如1,1-二苯乙烯不能聚合。

16
具体可分为以下几种情况: (1)取代基吸电子能力较弱,如偏二氯乙烯中的氯,两个氯吸电子作用的叠加,使单体更易聚合。 (2)取代基吸电子能力强,如偏二腈乙烯,两个腈基强吸电子作用使双键上电荷密度降低太多,从而使双键失去了与自由基加成的能力,只能阴离子聚合,而难自由基聚合。 偏二氯乙烯 偏二腈乙烯

17
(3)两个取代基都是给电子性,如异丁烯中的两个甲基,给电子作用的叠加,使异丁烯不能发生自由基聚合,而易于阳离子聚合。
(4)两个取代基中,一个是弱给电子性,另一个是强吸电子性,如甲基丙烯酸酯类,这类单体易发生自由基聚合反应。 甲基丙烯酸甲酯
两个取代基中,一个是弱给电子性,另一个是强吸电子性,如甲基丙烯酸酯类,这类单体易发生自由基聚合反应。 甲基丙烯酸甲酯.")
18
氟原子半径较小,仅大于氢原子,不会造成空间位阻。
2、 1,2双取代的烯类化合物XCH=CHY ,因结构对称,极 化程度低,位阻效应大,一般不能聚合或只能形成二聚 体。 3、 三取代、四取代的烯类化合物一般不能聚合,但氟代乙 烯例外。 例如:氟乙烯、1,1-二氟乙烯、1,2-二氟乙烯、三氟乙 烯、四氟乙烯均可聚合。 不论氟代的数量和位置,均极易聚合。 原因: 氟原子半径较小,仅大于氢原子,不会造成空间位阻。

19
3.3 自由基聚合机理 自由基聚合研究的两项重要指标: 聚合速率 分子量 自由基聚合的机理 聚合动力学

20
一、自由基聚合的基元反应 自由基:单电子物质,顺磁性,活性高,具有打开双键的能力。 自由基的活性:决定聚合速度
链引发 链增长 链终止 链转移反应

21
(一)链引发 链引发反应是形成单体自由基活性种的反应。 用引发剂引发时,有下列两步反应: 1、引发剂I分解,形成初级自由基 :
反应特征:吸热反应,活化能高,约105~150kJ/mol,反应速率小,分解速率常数约10-4~10-6s-1。

22
2、初级自由基与单体加成,形成单体自由基。
反应特征:放热反应,活化能低,约20~34kJ/mol,反应速率大,增长速率常数约102~ 。 链引发包含第二步。因为,虽然这一步反应与后继的链增长反应相似,但有一些副反应可以使某些初级自由基不参与单体自由基的形成,也就无法链增长。

23
(二)链增长 链增长反应:在链引发阶段形成的单体自由基不断地和单体分子结合生成链自由基的过程,实际上是加成反应。 反应特征:
1.放热反应,聚合热约55~95kJ/mol; 2. 增长活化能低,约20~34kJ/mol,增长速率极高,增长速率常数约102~ ,在0.01~几秒钟内,就可以使聚合度达到数千万,难以控制。 因此,在自由基聚合反应体系内,往往只存在单体和聚合物两部分,不存在聚合度递增的一系列中间产物。

24
自由基聚合反应中,结构单元间的连接存在“头-尾”、“头-头”(或“尾-尾”)两种可能的形式,一般以头-尾结构为主。
head-to-tail addition head-to-head addition 原因: (1)电子效应:头-尾连接时,自由基上的独电子与取代基构成共轭体系,使自由基稳定。而头头连接时无共轭效应,自由基不稳定。两者活化能相差34 ~ 42 kJ/mol。共轭稳定性较差的单体,容易出现头头结构。聚合温度升高,头头结构增多。 (2)位阻效应:以头-尾方式结合时,空间位阻要比头-头方式结合时的小,故有利于头尾结合。
电子效应:头-尾连接时,自由基上的独电子与取代基构成共轭体系,使自由基稳定。而头头连接时无共轭效应,自由基不稳定。两者活化能相差34 ~ 42 kJ/mol。共轭稳定性较差的单体,容易出现头头结构。聚合温度升高,头头结构增多。 (2)位阻效应:以头-尾方式结合时,空间位阻要比头-头方式结合时的小,故有利于头尾结合。")
25
实验证明,由于电子效应和空间位阻效应双重因素,都促使反应以头-尾连接为主;但还不能做到序列结构上的绝对规整性。由于链自由基是平面结构,在平面上下进攻的几率各为50%,因此,从立体结构看来,自由基聚合物分子链上取代基在空间的排布是无规的,所对应的聚合物往往是无定型的。

26
(三)链终止 链终止反应:在一定条件下,增长链自由基失去活性形成稳定聚合物分子的反应。 可分为偶合终止和歧化终止。
链终止 链终止反应:在一定条件下,增长链自由基失去活性形成稳定聚合物分子的反应。 可分为偶合终止和歧化终止。")
27
1、偶合终止 两链自由基的独电子相互结合成共价键的终止反应。 偶合终止所得大分子的特征: 大分子的聚合度为链自由基重复单元数的两倍;中间头-头结构。 若有引发剂引发聚合并无链转移反应,大分子两端均为引发剂残基。

28
2、歧化终止 某链自由基夺取另一链自由基相邻碳原子上的氢原子或其它原子的终止反应。 歧化终止所得大分子的特征: 大分子的聚合度与链自由基中单元数相同; 每个大分子只有一端为引发剂残基,另一端为饱和或不饱和结构,两者各半。

29
3、链终止反应特征: 活化能很低,只有8~21kJ/mol,甚至(偶合终止)为零;终止速率常数极高,约104~ ;链双基终止受扩散控制。 链终止方式与单体种类和聚合条件有关。一般而言,单体位阻大,聚合温度高,难以偶合终止,多以歧化终止为主。 例如:60℃以下苯乙烯聚合以几乎全为偶合终止, 60℃以 上歧化终止逐步增多。 60℃以下甲基丙烯酸甲酯聚合两种终 止方式均有, 60℃以上则以歧化终止逐步为主。 链增长和链终止是一对竞争反应。主要受反应速率常数和反应物质浓度的大小影响。 在自由基聚合的三步基元反应中,由于引发速率最小,是控制整个聚合速率的关键。

30
(四)链转移反应 链转移反应:链自由基从其它分子上夺取一个原子而终止成为稳定的大分子,而失去原子的分子又成为一个新的自由基,继续新链的增长,使聚合反应继续下去。 1、链转移反应有以下形式: (1)向溶剂或链转移剂转移 链自由基向溶剂分子转移的结果,使聚合度降低,聚合速率不变或稍有降低,视新生自由基的活性而定。

31
(2)向单体转移 链自由基将独电子转移到单体上,产生的单体自由基开始新的链增长,而链自由基本身因链转移提早终止,结果使聚合度降低,但转移后自由基数目并未减少,活性也未减弱,故聚合速率并不降低。向单体转移的速率与单体结构有关。如氯乙烯单体因C-Cl键能较弱而易于链转移。
向单体转移 链自由基将独电子转移到单体上,产生的单体自由基开始新的链增长,而链自由基本身因链转移提早终止,结果使聚合度降低,但转移后自由基数目并未减少,活性也未减弱,故聚合速率并不降低。向单体转移的速率与单体结构有关。如氯乙烯单体因C-Cl键能较弱而易于链转移。")
32
(3)向引发剂转移(也称为引发剂的诱导分解)
链自由基向引发剂转移,自由基数目并无增减,只是损失了一个引发剂分子;结果是反应体系中自由基浓度不变,聚合物分子量降低,引发剂效率下降。 以上所有链自由基向低分子物质转移的结果,都使聚合物的分子量降低;若新生自由基的活性不衰减,则不降低聚合速率。

33
链自由基可从已形成的“死”大分子上夺取原子而转移。
(4)向大分子转移 链自由基可从已形成的“死”大分子上夺取原子而转移。 向大分子转移一般发生在叔氢原子或氯原子上,结果使叔碳原子上带有独电子,形成大自由基,又进行链增长,形成支链高分子;或相互偶合成交联高分子。转化率高,聚合物浓度大时,容易发生这种转移。 支化
向大分子转移. 链自由基可从已形成的 死 大分子上夺取原子而转移。 向大分子转移一般发生在叔氢原子或氯原子上,结果使叔碳原子上带有独电子,形成大自由基,又进行链增长,形成支链高分子;或相互偶合成交联高分子。转化率高,聚合物浓度大时,容易发生这种转移。 支化.")
34
链转移反应不是自由基聚合必须经过的基元反应,但具有十分重要的意义。
2、阻聚作用 自由基向某些物质转移后,形成稳定的自由基,不能再引发单体聚合,只能与其它自由基双基终止;导致聚合过程停止。结果,体系中初期无聚合物形成,出现了所谓的“诱导期”,这种现象称为阻聚作用。 阻聚剂:具有阻聚作用的物质,如苯醌。 阻聚反应不是聚合的基元反应。 链转移反应不是自由基聚合必须经过的基元反应,但具有十分重要的意义。

35
二、自由基聚合反应的特征 1、自由基聚合反应由链的引发、增长、终止、转移等基元反应组成,其中引发速率最小,是控制总聚合速率的关键。慢引发、快增长、速终止。 2、只有链增长反应才使聚合度增加,自由基聚合时间短,反应混合物中仅由单体和聚合物组成;在聚合过程中,聚合度变化小。聚合度与聚合时间基本无关。 图1 自由基聚合过程中分子量与时间的关系

36
3、在聚合过程中,单体浓度逐步降低,聚合物浓度相应提高(见图2)。延长聚合时间主要是提高转化率,对分子量影响较小。凝胶效应将使分子量增大。
图2 自由基聚合过程中浓度与时间的关系 4、少量(0.01%~0.1%)阻聚剂可使自由基聚合反应终止。
阻聚剂可使自由基聚合反应终止。")
37
3.4 链引发反应 一、引发剂和引发作用 引发剂一般要求:
3.4 链引发反应 要产生自由基,最常用的方法是在聚合体系中引入引发剂,其次是采用热、光和高能辐射等方法。 一、引发剂和引发作用 引发剂一般要求: 容易分解成自由基的化合物,分子结构上具有弱键。 常用的引发剂有: 偶氮化合物、有机过氧化合物、无机过氧化合物和氧化-还原引发体系等。

38
(一)引发剂的种类 1、偶氮类引发剂 使用温度:45 ~ 65℃,解离能105kJ/mol。油溶性。
分解后形成的异丁腈自由基是碳自由基,缺乏脱氢能力,故不能作接枝聚合的引发剂。 分解反应只形成一种自由基, 无诱导分解; 比较稳定,能单独安全保存; 分解时有N2逸出

39
2、有机过氧类引发剂 最简单的过氧化物:过氧化氢。活化能较高,20kJ/mol, 一般不单独用作引发剂。 过氧化氢分子中一个氢原子被有机基团取代,称为“氢过 氧化物”,两个氢原子被取代,称为“过氧化物”。均可用作自 由基聚合引发剂。

40
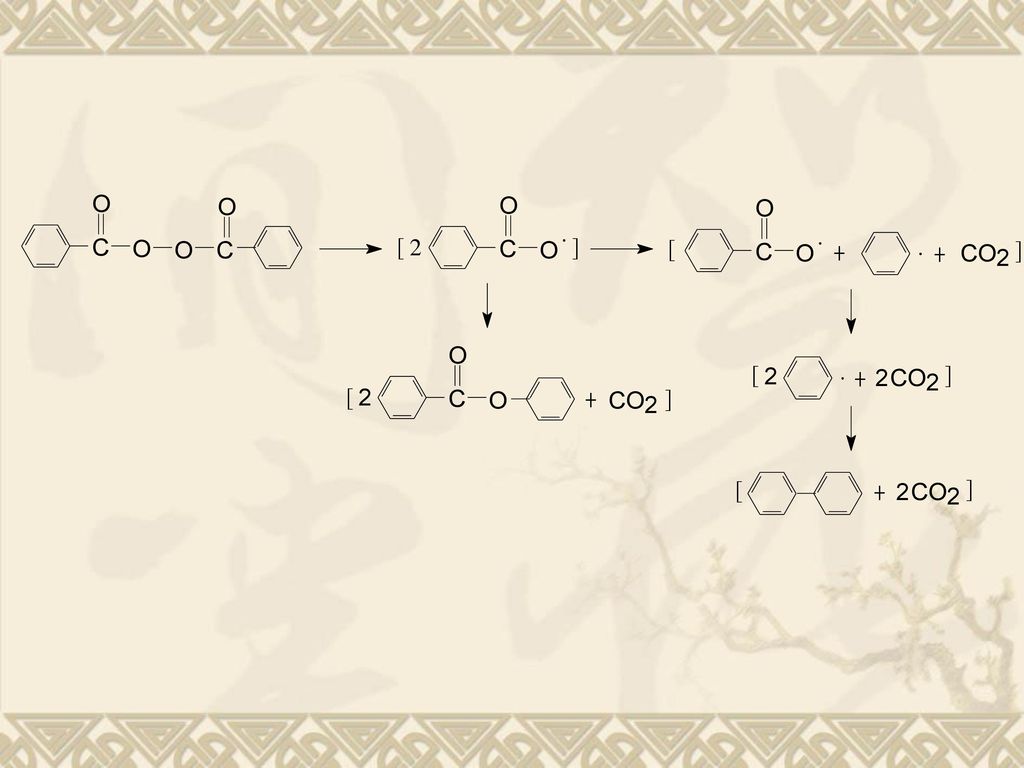
过氧化类引发剂的典型代表:过氧化二苯甲酰(BPO)。
BPO中O—O键部分的电子云密度大而相互排斥,容易断裂。 分解温度:60~80℃,解离能124kJ/mol。 BPO的分解分两步,第一步分解成苯甲酰自由基,第二 步分解成苯基自由基,并放出CO2。

41
3、无机过氧类引发剂 过硫酸盐,如过硫酸钾(K2S2O8) 过硫酸铵[(NH4)2S2O8]。 水溶性引发剂,主要用于乳液聚合和水溶液聚合。 分解温度:60~80℃,解离能109~140kJ/mol。
![3、无机过氧类引发剂 过硫酸盐,如过硫酸钾(K2S2O8) 过硫酸铵[(NH4)2S2O8]。 水溶性引发剂,主要用于乳液聚合和水溶液聚合。 分解温度:60~80℃,解离能109~140kJ/mol。](http://slidesplayer.com/slide/11130699/59/images/41/3%E3%80%81%E6%97%A0%E6%9C%BA%E8%BF%87%E6%B0%A7%E7%B1%BB%E5%BC%95%E5%8F%91%E5%89%82+%E8%BF%87%E7%A1%AB%E9%85%B8%E7%9B%90%EF%BC%8C%E5%A6%82%E8%BF%87%E7%A1%AB%E9%85%B8%E9%92%BE%EF%BC%88K2S2O8%EF%BC%89+%E8%BF%87%E7%A1%AB%E9%85%B8%E9%93%B5%5B%28NH4%292S2O8%5D%E3%80%82+%E6%B0%B4%E6%BA%B6%E6%80%A7%E5%BC%95%E5%8F%91%E5%89%82%EF%BC%8C%E4%B8%BB%E8%A6%81%E7%94%A8%E4%BA%8E%E4%B9%B3%E6%B6%B2%E8%81%9A%E5%90%88%E5%92%8C%E6%B0%B4%E6%BA%B6%E6%B6%B2%E8%81%9A%E5%90%88%E3%80%82+%E5%88%86%E8%A7%A3%E6%B8%A9%E5%BA%A6%EF%BC%9A60%EF%BD%9E80%E2%84%83%EF%BC%8C%E8%A7%A3%E7%A6%BB%E8%83%BD109%EF%BD%9E140kJ%2Fmol%E3%80%82.jpg "3、无机过氧类引发剂 过硫酸盐,如过硫酸钾(K2S2O8) 过硫酸铵[(NH4)2S2O8]。 水溶性引发剂,主要用于乳液聚合和水溶液聚合。 分解温度:60~80℃,解离能109~140kJ/mol。")
42
4、氧化-还原引发体系 将有机或无机过氧化物与还原剂复合,可组成氧化—还 原引发体系。 优点:活化能低(40~60kJ/mol),引发温度低(0~ 50℃),聚合速率大。 有水溶性和油溶性氧化—还原引发体系之分。前者用于 乳液聚合和水溶液聚合,后者用于溶液聚合和本体聚合。 反应机理:直接电荷转移或先形成中间络合物。

43
(1)水溶性氧化-还原引发体系 氧化剂:过氧化氢、过硫酸盐、氢过氧化物等; 还原剂:无机还原剂(Fe2+、Cu+、NaHSO3、 NaS2O3等) 和有机还原剂(醇、胺、草酸等)。
水溶性氧化-还原引发体系 氧化剂:过氧化氢、过硫酸盐、氢过氧化物等; 还原剂:无机还原剂(Fe2+、Cu+、NaHSO3、 NaS2O3等) 和有机还原剂(醇、胺、草酸等)。")
44
组成氧化—还原体系后,分解活化能大大降低。
例如: 过氧化氢:220kJ/mol;过氧化氢+亚铁盐:40kJ/mol 过硫酸钾:140kJ/mol;过硫酸钾+亚铁盐:50kJ/mol 异丙苯过氧化氢:125kJ/mol;异丙苯过氧化氢+亚铁 盐:50kJ/mol 还原剂用量一般应较氧化剂少,否则还原剂进一步与自 由基反应,使活性消失。 若还原剂过量:

45
亚硫酸盐和硫代硫酸盐常与过硫酸盐构成氧化—还原体系,形成两个自由基。

46
(2)油溶性氧化-还原引发体系 氧化剂:氢过氧化物、过氧化二烷基、过氧化二酰基; 还原剂:叔胺、环烷酸盐、硫醇、有机金属化合物 (Al(C2H5)3、B(C2H5)3等)。 该氧化-还原引发体系较单纯的BPO引发剂具有大的多的分解速率常数。
3、B(C2H5)3等)。 该氧化-还原引发体系较单纯的BPO引发剂具有大的多的分解速率常数。 .")
47
(二)引发剂分解动力学 研究引发剂浓度与时间、温度间的定量关系。 1、分解速率常数及半衰期 (1)分解速率常数 引发剂分解属于动力学一级反应,即分解速率Rd与引发剂浓度[I]的一次方成正比,微分式如下: kd—分解速率常数,单位为s-1、min-1或h-1。
![(二)引发剂分解动力学 研究引发剂浓度与时间、温度间的定量关系。 1、分解速率常数及半衰期. (1)分解速率常数. 引发剂分解属于动力学一级反应,即分解速率Rd与引发剂浓度[I]的一次方成正比,微分式如下:](http://slidesplayer.com/slide/11130699/59/images/47/%EF%BC%88%E4%BA%8C%EF%BC%89%E5%BC%95%E5%8F%91%E5%89%82%E5%88%86%E8%A7%A3%E5%8A%A8%E5%8A%9B%E5%AD%A6+%E7%A0%94%E7%A9%B6%E5%BC%95%E5%8F%91%E5%89%82%E6%B5%93%E5%BA%A6%E4%B8%8E%E6%97%B6%E9%97%B4%E3%80%81%E6%B8%A9%E5%BA%A6%E9%97%B4%E7%9A%84%E5%AE%9A%E9%87%8F%E5%85%B3%E7%B3%BB%E3%80%82+1%E3%80%81%E5%88%86%E8%A7%A3%E9%80%9F%E7%8E%87%E5%B8%B8%E6%95%B0%E5%8F%8A%E5%8D%8A%E8%A1%B0%E6%9C%9F.+%EF%BC%881%EF%BC%89%E5%88%86%E8%A7%A3%E9%80%9F%E7%8E%87%E5%B8%B8%E6%95%B0.+%E5%BC%95%E5%8F%91%E5%89%82%E5%88%86%E8%A7%A3%E5%B1%9E%E4%BA%8E%E5%8A%A8%E5%8A%9B%E5%AD%A6%E4%B8%80%E7%BA%A7%E5%8F%8D%E5%BA%94%EF%BC%8C%E5%8D%B3%E5%88%86%E8%A7%A3%E9%80%9F%E7%8E%87Rd%E4%B8%8E%E5%BC%95%E5%8F%91%E5%89%82%E6%B5%93%E5%BA%A6%5BI%5D%E7%9A%84%E4%B8%80%E6%AC%A1%E6%96%B9%E6%88%90%E6%AD%A3%E6%AF%94%EF%BC%8C%E5%BE%AE%E5%88%86%E5%BC%8F%E5%A6%82%E4%B8%8B%EF%BC%9A.jpg "kd—分解速率常数,单位为s-1、min-1或h-1。")
48
将上式积分,得: [I]0—引发剂的起始浓度,单位为mol/L。 [I] —时间为t时的引发剂浓度,单位为mol/L。 固定温度,测定不同时间t下的引发剂浓度变化,以ln ([I]/[I]0)对t作图,由斜率可求出引发剂的分解速率常数kd。
![将上式积分,得: [I]0—引发剂的起始浓度,单位为mol/L。 [I] —时间为t时的引发剂浓度,单位为mol/L。 固定温度,测定不同时间t下的引发剂浓度变化,以ln ([I]/[I]0)对t作图,由斜率可求出引发剂的分解速率常数kd。](http://slidesplayer.com/slide/11130699/59/images/48/%E5%B0%86%E4%B8%8A%E5%BC%8F%E7%A7%AF%E5%88%86%EF%BC%8C%E5%BE%97%EF%BC%9A+%5BI%5D0%E2%80%94%E5%BC%95%E5%8F%91%E5%89%82%E7%9A%84%E8%B5%B7%E5%A7%8B%E6%B5%93%E5%BA%A6%EF%BC%8C%E5%8D%95%E4%BD%8D%E4%B8%BAmol%2FL%E3%80%82+%5BI%5D+%E2%80%94%E6%97%B6%E9%97%B4%E4%B8%BAt%E6%97%B6%E7%9A%84%E5%BC%95%E5%8F%91%E5%89%82%E6%B5%93%E5%BA%A6%EF%BC%8C%E5%8D%95%E4%BD%8D%E4%B8%BAmol%2FL%E3%80%82+%E5%9B%BA%E5%AE%9A%E6%B8%A9%E5%BA%A6%EF%BC%8C%E6%B5%8B%E5%AE%9A%E4%B8%8D%E5%90%8C%E6%97%B6%E9%97%B4t%E4%B8%8B%E7%9A%84%E5%BC%95%E5%8F%91%E5%89%82%E6%B5%93%E5%BA%A6%E5%8F%98%E5%8C%96%EF%BC%8C%E4%BB%A5ln+%28%5BI%5D%2F%5BI%5D0%29%E5%AF%B9t%E4%BD%9C%E5%9B%BE%EF%BC%8C%E7%94%B1%E6%96%9C%E7%8E%87%E5%8F%AF%E6%B1%82%E5%87%BA%E5%BC%95%E5%8F%91%E5%89%82%E7%9A%84%E5%88%86%E8%A7%A3%E9%80%9F%E7%8E%87%E5%B8%B8%E6%95%B0kd%E3%80%82.jpg "将上式积分,得: [I]0—引发剂的起始浓度,单位为mol/L。 [I] —时间为t时的引发剂浓度,单位为mol/L。 固定温度,测定不同时间t下的引发剂浓度变化,以ln ([I]/[I]0)对t作图,由斜率可求出引发剂的分解速率常数kd。")
49
(2)半衰期 工程上常将一级反应的反应速率用半衰期表示。 半衰期— 指引发剂分解至起始浓度一半所需的时间, 以t1/2表示,单位通常为h。 分解速率常数和半衰期是表示引发剂活性的两个物理量,分解速率常数愈大,或半衰期愈短,引发剂的活性愈高。
半衰期 工程上常将一级反应的反应速率用半衰期表示。 半衰期— 指引发剂分解至起始浓度一半所需的时间, 以t1/2表示,单位通常为h。 分解速率常数和半衰期是表示引发剂活性的两个物理量,分解速率常数愈大,或半衰期愈短,引发剂的活性愈高。")
50
实际应用时,常选择半衰期与聚合时间相当的引发剂。
半衰期用来表示其活性的大小。 t1/2 ≥ 6h,低活性 t1/2 ≤ 1h,高活性 6h > t1/2 > 1h,中等活性 实际应用时,常选择半衰期与聚合时间相当的引发剂。

51
2、分解速率常数与温度间的关系 引发剂的分解速率常数与温度关系遵循Arrhenius经验公式: 或
Ed — 分解活化能;Ad — 频率因子。 常用引发剂的kd约10-4~10-6s-1,Ed约105~150kJ/mol,单个分子的Ad一般在1013~1014。 由于Ed为正值,随温度升高, kd增大。

52
(三)引发剂效率 聚合体系中的引发剂并不是全部分解可以引发聚合的,还有一部分引发剂由于诱导分解和/或笼蔽效应伴随的副反应而损耗,因此,需引入引发剂效率的概念。 引发剂效率 f — 引发聚合的部分引发剂占引发 剂分解或消耗总量的分率。

53
1、诱导分解 诱导分解实际上是自由基向引发剂的转移反应。尤其是过氧化合物引发剂。
转移的结果是链自由基夺取引发剂分子中的一部分,变成了稳定分子,剩余的引发剂残片形成自由基。整个过程中自由基数量没有增加,但消耗了一个引发剂分子,从而使引发剂效率降低。 氢过氧化物也容易发生诱导分解。

54
2、笼蔽效应及其伴随的副反应 笼蔽效应—当体系中引发剂浓度较低时,引发剂分子处于 单体或溶剂的包围中而不能发挥作用,
自由基在单体或溶剂的“笼子”中的的平均寿命很短,如来 不及扩散出笼子,就可能发生副反应,形成稳定分子。 结果是消耗了引发剂,降低了引发剂效率。

56
(1)引发剂本身的影响:偶氮类引发剂(如AIBN)一般无诱导分解,而过氧类引发剂(如BPO)易发生诱导分解,使f 值下降。
单体的活性:若单体具有较高的活性,能迅速与自由基作用,引发增长,因此引发剂效率较高;若单体的活性较低,对自由基的捕捉能力较弱,为诱导分解创造条件,则引发剂效率低。 表2 AIBN引发不同烯类单体的引发剂效率f (%)
")
57
3、引发剂的选择 (1)首先根据聚合方法选择引发剂类型。 本体、悬浮和溶液聚合:选用油溶性引发剂 乳液聚合和水溶液聚合:选用水溶性引发剂
首先根据聚合方法选择引发剂类型。 本体、悬浮和溶液聚合:选用油溶性引发剂 乳液聚合和水溶液聚合:选用水溶性引发剂")
58
一般选择在聚合温度下半衰期为5-10h左右的引发剂。
(2)根据聚合温度选择活化能或半衰期适当的引发剂,使自由基形成速率和聚合速率适中。 引发剂在不同温度下有不同的半衰期。 半衰期过长,分解速率低,聚合时间长; 半衰期过短,则引发剂在早期大量分解,易引起爆聚,后期则无足够的引发剂维持适当的聚合速率。 一般选择在聚合温度下半衰期为5-10h左右的引发剂。 (3)其他 毒性、使用场合、产品质量、安全生产
根据聚合温度选择活化能或半衰期适当的引发剂,使自由基形成速率和聚合速率适中。 引发剂在不同温度下有不同的半衰期。 半衰期过长,分解速率低,聚合时间长; 半衰期过短,则引发剂在早期大量分解,易引起爆聚,后期则无足够的引发剂维持适当的聚合速率。 一般选择在聚合温度下半衰期为5-10h左右的引发剂。 (3)其他. 毒性、使用场合、产品质量、安全生产.")
59
3.5 聚合速率 一、概述 聚合动力学研究的主要内容: 聚合速率和分子量 聚合速率的变化表示:常用转化率-时间曲线 1. 诱导期
3.5 聚合速率 一、概述 聚合动力学研究的主要内容: 聚合速率和分子量 聚合速率的变化表示:常用转化率-时间曲线 1. 诱导期 2. 聚合初期 3. 聚合中期 4. 聚合后期 自由基聚合的转化率-时间关系

60
聚合速率的测定方法: 1、直接法 加入沉淀剂使聚合物沉淀,或蒸馏出单体,使聚合中断,然后经分离、精制、干燥、称重等程序,求出聚合物的质量。
2、间接法 测定聚合过程中比容、粘度、折光率、吸收光谱等物理性质的变化,间接求出聚合物量,从而可得到聚合速率。最常用的是比容的测定——膨胀计法。

61
二、自由基聚合微观动力学 1、链引发 (1)引发剂分解成初级自由基: kd I 2R. (2)初级自由基同单体加成形成单体自由基: k1
+ M RM. 引发速率一般仅决定于初级自由基的生成速率,而与单体浓度无关。

62
引发速率(即初级自由基的生成速率)Ri:
Ri = d[R.] / dt = 2kd[I] 由于诱导分解和/或笼蔽效应伴随的副反应,初级自由基或分解的引发剂并不全部参加引发反应,故需引入引发剂效率f 。 Ri = d[R.] / dt = 2 f kd[I] 式中: I— 引发剂; M— 单体; R.— 初级自由基; k— 速率常数。 [ ]— 浓度; d — 分解 i — 引发 kd :10-4~10-6s-1; f :0.6~0.8; Ri :10-8~10-10mol/(L.s)
")
63
2、链增长 … 链增长反应是RM·连续加上单体分子的反应。 RM. +M kP1 RM2. kP2 kP3 RM3. RMX.
(1)推导自由基聚合动力学的假定: 假定一:等活性理论假定 链自由基的活性与链长基本无关,各步速率常数相等, kP1=kP2=kP3=kP4= … kPx= kP
推导自由基聚合动力学的假定: 假定一:等活性理论假定. 链自由基的活性与链长基本无关,各步速率常数相等, kP1=kP2=kP3=kP4= … kPx= kP.")
64
令自由基浓度[M.]代表大小不等的自由基RM.、 RM2.、RM3.、 … RMX.浓度的总和,则链增长速率方程可写成:
(6) 由于:kp :102~104L/(mol . s); [M.] :10-7~10-9mol/L; [M]: 1~10mol/L;故 Rp :10-4~10-6mol/(L.s)
![令自由基浓度[M.]代表大小不等的自由基RM.、 RM2.、RM3.、 … RMX.浓度的总和,则链增长速率方程可写成:](http://slidesplayer.com/slide/11130699/59/images/64/%E4%BB%A4%E8%87%AA%E7%94%B1%E5%9F%BA%E6%B5%93%E5%BA%A6%5BM.%5D%E4%BB%A3%E8%A1%A8%E5%A4%A7%E5%B0%8F%E4%B8%8D%E7%AD%89%E7%9A%84%E8%87%AA%E7%94%B1%E5%9F%BARM.%E3%80%81+RM2.%E3%80%81RM3.%E3%80%81+%E2%80%A6+RMX.%E6%B5%93%E5%BA%A6%E7%9A%84%E6%80%BB%E5%92%8C%EF%BC%8C%E5%88%99%E9%93%BE%E5%A2%9E%E9%95%BF%E9%80%9F%E7%8E%87%E6%96%B9%E7%A8%8B%E5%8F%AF%E5%86%99%E6%88%90%EF%BC%9A.jpg "(6) 由于:kp :102~104L/(mol . s); [M.] :10-7~10-9mol/L; [M]: 1~10mol/L;故 Rp :10-4~10-6mol/(L.s)")
65
3、链终止 链终止速率— 自由基消失速率,以Rt表示。 偶合终止: 歧化终止: 终止总速率:
在以上三式中,系数2表示每一次终止反应消失两个自由基,为美国习惯,欧洲习惯一般无系数2。

66
(2)假定二:稳态假定 在聚合过程中,链增长的过程并不改变自由基的浓度,链引发和链终止这两个相反的过程在某一时刻达到平衡,体系处于“稳定状态” ;或者说引发速率和终止速率相等,Ri=Rt,构成动态平衡。
假定二:稳态假定 在聚合过程中,链增长的过程并不改变自由基的浓度,链引发和链终止这两个相反的过程在某一时刻达到平衡,体系处于 稳定状态 ;或者说引发速率和终止速率相等,Ri=Rt,构成动态平衡。")
67
4、聚合总速率的推导: I 聚合总速率通常以单体消耗速率(-d[M]/dt)表示。 (3)假定三:聚合度很大的假定
基元反应中,链引发和链增长这两步都消耗单体,对于高分子,聚合度很大,用于引发的单体远远少于增长消耗的单体,因此引入第三个假定。 增长速率远远大于引发速率,Rp≥Ri, 聚合总速率就等于链增长速率。
![4、聚合总速率的推导: I 聚合总速率通常以单体消耗速率(-d[M]/dt)表示。 (3)假定三:聚合度很大的假定](http://slidesplayer.com/slide/11130699/59/images/67/4%E3%80%81%E8%81%9A%E5%90%88%E6%80%BB%E9%80%9F%E7%8E%87%E7%9A%84%E6%8E%A8%E5%AF%BC%EF%BC%9A+I+%E8%81%9A%E5%90%88%E6%80%BB%E9%80%9F%E7%8E%87%E9%80%9A%E5%B8%B8%E4%BB%A5%E5%8D%95%E4%BD%93%E6%B6%88%E8%80%97%E9%80%9F%E7%8E%87%EF%BC%88-d%5BM%5D%2Fdt%EF%BC%89%E8%A1%A8%E7%A4%BA%E3%80%82+%EF%BC%883%EF%BC%89%E5%81%87%E5%AE%9A%E4%B8%89%EF%BC%9A%E8%81%9A%E5%90%88%E5%BA%A6%E5%BE%88%E5%A4%A7%E7%9A%84%E5%81%87%E5%AE%9A.jpg "基元反应中,链引发和链增长这两步都消耗单体,对于高分子,聚合度很大,用于引发的单体远远少于增长消耗的单体,因此引入第三个假定。 增长速率远远大于引发速率,Rp≥Ri, 聚合总速率就等于链增长速率。")
68
总聚合速率的普适方程 II、引发剂引发的自由基聚合反应的总聚合速率为:

69
(a)对引发剂浓度1/2次方的偏离: 聚合速率与引发剂浓度1/2次方成正比是双基终止的结果;若是单基终止(对引发剂浓度的反应级数为1级),如存在凝胶效应或沉淀聚合时,链自由基活性末端受到包埋,难以双基终止,往往是单基终止和双基终止并存,则聚合速率对引发剂浓度的反应级数介于0.5~1之间。可表示为: 0.5级—双基终止时的引发剂浓度的反应级数; 1级— 单基终止时的引发剂浓度的反应级数。

70
(b)对单体浓度一次方的偏离: 若初级自由基与单体的引发反应较慢,与引发剂的分解速率相当,链引发速率则与单体浓度有关,应表示为:
对单体浓度一次方的偏离: 若初级自由基与单体的引发反应较慢,与引发剂的分解速率相当,链引发速率则与单体浓度有关,应表示为:")
71
V、适合于各种情况的聚合速率表达式: 通常,式中: n=0.5~1.0;m=1~1.5(个别可达2)
")
72
三、自由基聚合基元反应速率常数 综合常用单体的自由基聚合,各基元反应的速率常数等参数可归纳如下:
kd = 10-4~ 10-6, kp = 102~ 104, kt = 106~ 108 Ed=105~150 kJ/mol,Ep=16~33 kJ/mol,Et=8~21 kJ/mol 增长速率:Rp = kp[M][M.] = 10-4~10-6 mol/L. s 终止速率:Rt = kt[M.]2 = 10-8~10-10 mol/L. s 因此最终可得高分子,聚合度为103 ~105。 总聚合速率由最慢的引发反应来控制。

73
四、温度对聚合速率的影响 k=Ae-E/RT 总聚合速率常数k与温度T(K)遵循Arrhenius经验公式:
总活化能E=(Ep-Et/2)+Ed/2 通常,Ed≈125 kJ/mol, Ep≈29 kJ/mol, Et≈17 kJ/mol, 总活化能E约为83kJ/mol,为正值,表明温度升高,速率常数k增大。
+Ed/2. 通常,Ed≈125 kJ/mol, Ep≈29 kJ/mol, Et≈17 kJ/mol, 总活化能E约为83kJ/mol,为正值,表明温度升高,速率常数k增大。")
74
一聚合体系,T1 = 50℃,T2 = 60 ℃, E = 80 kJ/mol,R = J/mol.K, 求 k2/k1。 解:

75
体系一:K2S2O Ed = 140 kJ/mol 体系二:K2S2O8—Fe Ed = 50 kJ/mol T = 60℃,R = J/mol.K,求k2/k1。 解:Ep 取 29 kJ/mol, Et 取 17 kJ/mol,则

76
选择Ed较低的引发剂,则反应速率的提高要比升高T更显著。
氧化—还原引发体系能在较低温度下保持较高聚合速率,就是此理。

77
五、自动加速现象 引发剂引发的自由基聚合反应的总聚合速率为:
从式(13)可看出,聚合总速率与引发剂浓度平方根、单体浓度一次方根成正比,因此,随聚合速率增加,单体浓度和引发剂浓度下降,聚合总速率理应降低;但达到一定转化率(如15%~20%)后,聚合速率会大幅度上升。这种现象,称为自动加速现象。直到后期,聚合速率才逐渐减慢。自由基聚合的转化率-时间曲线往往呈S形。
可看出,聚合总速率与引发剂浓度平方根、单体浓度一次方根成正比,因此,随聚合速率增加,单体浓度和引发剂浓度下降,聚合总速率理应降低;但达到一定转化率(如15%~20%)后,聚合速率会大幅度上升。这种现象,称为自动加速现象。直到后期,聚合速率才逐渐减慢。自由基聚合的转化率-时间曲线往往呈S形。")
78
甲基丙烯酸甲酯聚合转化率-时间曲线 溶剂:苯,T=50℃,引发剂:BPO (曲线上数字表示MMA的百分浓度)
")
79
1、 自动加速现象主要是由体系的粘度增加引起的,因此又称凝胶效应。可用扩散控制理论和自由基双基终止机理来解释。
2、自动加速现象产生的原因: 链终止反应受扩散控制所致。 (1)链自由基的双基终止过程的三步曲: 链自由基的平移; 链段重排,使活性中心靠近; 双基相互反应而使链终止。
链自由基的双基终止过程的三步曲: 链自由基的平移; 链段重排,使活性中心靠近; 双基相互反应而使链终止。")
80
(2)随转化率增加,kp/kt1/2综合值的变化:
聚合初期,体系粘度较低,聚合正常。 随转化率增加,粘度上升,链段重排受阻,双基终止困难,kt下降。但单体运动不受影响,kp影响不大。 据测定,当转化率大40%~50%时, kt下降达上百倍。因此kp/ kt1/2增加近10倍,活性链寿命也增长10多倍。导致聚合速率和分子量大幅度上升。 转化率继续上升后,粘度增大至单体的运动也受阻,则 kt和kp都下降, kp/kt1/2 综合值减小,聚合总速率下降。最后甚至停止反应。

81
(3)链自由基卷曲、包埋的程度以及聚合速率的大小受聚合物在单体或溶剂中溶解性能的好坏的影响。通常,自动加速现象在良溶剂中较少出现,在非溶剂(沉淀剂)中出现得早、显著,在不良溶剂中自动加速现象介于以上两种情况。
链自由基卷曲、包埋的程度以及聚合速率的大小受聚合物在单体或溶剂中溶解性能的好坏的影响。通常,自动加速现象在良溶剂中较少出现,在非溶剂(沉淀剂)中出现得早、显著,在不良溶剂中自动加速现象介于以上两种情况。")
82
(1~3—采用非溶剂;4~5—采用不良溶剂;8~10—采用良溶剂)
11 溶剂对MMA聚合时自动加 速现象的影响 1— 硬脂酸丁酯 — 苯 2— 庚烷 — 氯仿 3— 环己烷 — 二氯甲烷 4— 醋酸正戊酯 — 本体聚合 5— 戊基氯 6— 醋酸乙酯 7— 四氯化碳 (1~3—采用非溶剂;4~5—采用不良溶剂;8~10—采用良溶剂)
")
83
六、聚合过程中速率变化的类型 1、转化率-时间曲线呈S形 任何单体的聚合,都可看成正常的聚合与自动加速现象叠加而成。
正常的聚合速率随单体转化率上升而降低;自动加速时的聚合速率则随单体转化率上升而上升。根据自动加速现象出现的时间和程度,叠加情况可分为三类。 1、转化率-时间曲线呈S形 聚合总速率的变化规律为:初期慢,中期加速,后期又转慢; 正常的聚合速率的降低不及自动加速引起的聚合速率上升。总的表现为加速。采用低活性引发剂(BPO、AIBN)时一般属此类。
时一般属此类。")
84
正常聚合与自动加速互补。选用半衰期略小于聚合时间的 引发剂,可实现匀速聚合。如氯乙烯聚合时,采用t1/2 = 2h的
2 匀速聚合 正常聚合与自动加速互补。选用半衰期略小于聚合时间的 引发剂,可实现匀速聚合。如氯乙烯聚合时,采用t1/2 = 2h的 过氧化碳酸酯类引发剂,基本属于此类。 3 前快后慢型 采用高活性引发剂时,聚合前期大量自由基产生,聚合速 率较大。中后期因引发剂减少,聚合速率降低,以致无法用 自动加速效应来弥补。如采用过氧化乙酰基环己烷磺酰作引 发剂是即属此类。可通过与低活性引发剂混用或后补加引发 剂来解决。 转化率-时间曲线 —常见S形 2—匀速反应; 3—前快后慢)
")
85
3.6 分子量和链转移反应 一、无链转移时的分子量 聚合速率 分子量 自由基聚合研究的两项重要指标:
3.6 分子量和链转移反应 一、无链转移时的分子量 自由基聚合研究的两项重要指标: 聚合速率 分子量 聚合速率和分子量随引发剂浓度、聚合温度等因素的改变而变化的规律相反。 聚合温度 引发剂浓度 聚合速率 分子量

86
1、动力学链长和聚合度 (1)动力学链长υ的定义: 每个活性种从引发阶段到终止阶段所消耗的单体分子数。
无链转移时,动力学链长为增长速率和引发速率的比。依据稳态时引发速率等于终止速率,则动力学链长可表示为增长速率与终止速率的比:

87
若自由基聚合反应由引发剂引发时,引发速率Ri = 2 f kd[I],则:
动力学链长与引发剂浓度平方根成反比。 因此,在实施自由基聚合时,增加引发剂提高聚合速率的措施,往往使产物聚合度下降。
![若自由基聚合反应由引发剂引发时,引发速率Ri = 2 f kd[I],则:](http://slidesplayer.com/slide/11130699/59/images/87/%E8%8B%A5%E8%87%AA%E7%94%B1%E5%9F%BA%E8%81%9A%E5%90%88%E5%8F%8D%E5%BA%94%E7%94%B1%E5%BC%95%E5%8F%91%E5%89%82%E5%BC%95%E5%8F%91%E6%97%B6%EF%BC%8C%E5%BC%95%E5%8F%91%E9%80%9F%E7%8E%87Ri+%3D+2+f+kd%5BI%5D%EF%BC%8C%E5%88%99%EF%BC%9A.jpg "动力学链长与引发剂浓度平方根成反比。 因此,在实施自由基聚合时,增加引发剂提高聚合速率的措施,往往使产物聚合度下降。")
88
(2)平均聚合度 与动力学链长υ的关系: 双基偶合终止时, 歧化终止时, 两种终止方式兼有时,υ< < 2υ 或: 式中C, D— 分别代表偶合终止和歧化终止的分率。
平均聚合度 与动力学链长υ的关系: 双基偶合终止时, 歧化终止时, 两种终止方式兼有时,υ< < 2υ 或: 式中C, D— 分别代表偶合终止和歧化终止的分率。")
89
2、聚合温度对聚合度的影响 温度上升,聚合速率上升,但聚合度下降。 E`=(Ep-Et/2)-Ed/2
Ed≈125 kJ/mol,Ep≈29 kJ/mol,Et≈17kJ/mol, 由Ep、 Et和Ed的大小可以得到综合活化能E`约为-41kJ/mol,为负值,表明温度升高,k`值或聚合度降低。 温度上升,聚合速率上升,但聚合度下降。

90
一聚合体系,T1 = 50℃,T2 = 60 ℃,E’ = -42 kJ/mol,
R = J/mol.K,求 k’1/k’2。 解:

91
二、链转移反应 1、概述 在自由基聚合体系中,若存在容易被夺去原子(如氢、氯等)的物质时,容易发生链转移反应。
链转移的结果,原来自由基终止,聚合度下降;新形成的自由基如有足够的活性,可以再引发体系中的单体分子反应,继续链增长。 式中,ktr— 链转移速率常数,ka为再引发速率常数。

92
(1)新自由基的活性与原自由基活性相同,聚合速率不变;
(2)新自由基活性减弱,出现缓聚现象; (3)新自由基没有引发活性,聚合停止,表现为阻聚作用。 实际生产中,链转移反应常被用来调节和控制分子量,如丁苯橡胶生产中用硫醇来调节分子量等。
新自由基活性减弱,出现缓聚现象; (3)新自由基没有引发活性,聚合停止,表现为阻聚作用。 实际生产中,链转移反应常被用来调节和控制分子量,如丁苯橡胶生产中用硫醇来调节分子量等。")
93
2、链转移反应对聚合度的影响 (1)活性链分别向单体、引发剂、溶剂等低分子物质发生链转移的反应式和速率方程:
活性链分别向单体、引发剂、溶剂等低分子物质发生链转移的反应式和速率方程:")
94
(2)存在链转移反应时,动力学链长和聚合度的重新定义:
动力学链长— 每个初级自由基自链引发开始到活性中心真正死亡止(不论双基终止,或单基终止,但不包括链转移终止)所消耗的单体分子总数。链转移时,活性中心并没有消失,因此动力学链长没有终止。 聚合度— 由于链转移反应导致聚合度的下降,因此,研究高分子的聚合度时要考虑聚合过程中存在的链转移反应,即须考虑真正终止(双基终止、单基终止)和链转移终止两种链终止方式。
所消耗的单体分子总数。链转移时,活性中心并没有消失,因此动力学链长没有终止。 聚合度— 由于链转移反应导致聚合度的下降,因此,研究高分子的聚合度时要考虑聚合过程中存在的链转移反应,即须考虑真正终止(双基终止、单基终止)和链转移终止两种链终止方式。")
95
双基终止反应暂且仅考虑歧化终止,则平均聚合度可表示为:增长速率与形成大分子的所有终止速率(包括链转移终止)之比:
Rp = kp[M•][M]; Rt = 2kt[M•]2 RtrM = ktrM[M•][M]; RtrI =ktrI[M•][I]; RtrS = ktrS[M•][S]

96
链转移常数C— 链转移速率常数与链增长速率常数之比,代表两种反应竞争能力的大小,表示为ktr/kp。
向单体、引发剂、溶剂的链转移速率常数CM、CI、 CS的定义: 右边四项分别代表正常聚合、向单体转移、向引发剂转 移、向溶剂转移对平均聚合度的贡献。

97
提高单体浓度有利于提高产物聚合度; 增大引发剂浓度会降低产物聚合度; 加入链转移剂降低产物聚合度; 此外产物聚合度还与单体、引发剂及链转移剂的链转移能力有关。

98
(i)常见单体的CM一般较小,多为10-5数量级,故可忽略。
例外:氯乙烯的CM较大,其聚合产物的分子量主要取决于CM,并且CM随温度的上升而增加,因此氯乙烯聚合中主要通过控制温度来调节聚合产物分子量,要得到高分子量的聚合物,聚合温度不能太高,一般不高于60oC。聚合速率的调节通过改变引发剂浓度来实现。 (ii)CI虽然比CM和CS大,但由于引发剂浓度一般很小,所以向引发剂转移造成产物聚合度下降的影响不大。 (iii)溶剂链转移常数CS取决于溶剂的结构,如分子中有活泼氢或卤原子时,CS一般较大。特别是脂肪族的硫醇CS较大,常用做分子量调节剂。
CI虽然比CM和CS大,但由于引发剂浓度一般很小,所以向引发剂转移造成产物聚合度下降的影响不大。 (iii)溶剂链转移常数CS取决于溶剂的结构,如分子中有活泼氢或卤原子时,CS一般较大。特别是脂肪族的硫醇CS较大,常用做分子量调节剂。")
99
3、向单体转移 进行本体聚合并采用偶氮类引发剂时,或存在溶剂而溶剂的链转移常数很小以至于可以忽略,链转移反应可以只考虑向单体转移的反应,大分子的平均聚合度则可以表示为下式: 按1/ Xn ~ Rp作图,由截距可求出向单体的链转移常数CM 。

100
若单体分子结构中含有键合力较小的原子,如叔氢原子、氯原子等,容易被自由基所夺取而发生链转移反应。
(1)单体结构对CM大小的影响 若单体分子结构中含有键合力较小的原子,如叔氢原子、氯原子等,容易被自由基所夺取而发生链转移反应。 表1 向单体的链转移常数CM×104 单体 温度 /℃ 30 50 60 70 80 甲基丙烯酸甲酯 丙烯腈 苯乙烯 醋酸乙烯酯 氯乙烯 0.12 0.15 0.32 0.94* 6.25 0.27 0.62 1.29 13.5 0.18 0.30 0.85 1.91 20.2 0.3 1.16 23.8 0.4 同一种单体,温度越高,越容易发生向单体转移。
单体结构对CM大小的影响. 若单体分子结构中含有键合力较小的原子,如叔氢原子、氯原子等,容易被自由基所夺取而发生链转移反应。 表1 向单体的链转移常数CM×104. 单体. 温度 /℃ 甲基丙烯酸甲酯. 丙烯腈. 苯乙烯. 醋酸乙烯酯. 氯乙烯 * 同一种单体,温度越高,越容易发生向单体转移。")
101
I、从表1中数据可见,如苯乙烯、甲基丙烯酸甲酯等单体的链转移常数较小,约10-4~10-5,对分子量并无严重影响。原因是C— H键较强,H不易被夺取。
II、醋酸乙烯酯的CM较大,主要是乙酰氧基上的甲基氢易被夺取,如下式所示:

102
III、由于C— Cl 键弱,Cl 易被夺取,因此,氯乙烯的CM值较高,约10-3 ,其转移速率远远超出了正常的终止速率,即Rtr
III、由于C— Cl 键弱,Cl 易被夺取,因此,氯乙烯的CM值较高,约10-3 ,其转移速率远远超出了正常的终止速率,即Rtr.M>Rt,因此,聚氯乙烯的平均聚合度主要决定于向单体转移常数。

103
(2)温度对CM大小的影响 CM是两个速率常数的比值,因此可用Arrhenius经验公式讨论温度对链转移常数的影响。 对于氯乙烯单体的聚合,向氯乙烯链转移常数CM与温度有如下指数关系: 转移活化能和增长活化能的差值30.5kJ/mol,为正值,表明温度升高,CM值增加,聚合度降低。
温度对CM大小的影响 CM是两个速率常数的比值,因此可用Arrhenius经验公式讨论温度对链转移常数的影响。 对于氯乙烯单体的聚合,向氯乙烯链转移常数CM与温度有如下指数关系: 转移活化能和增长活化能的差值30.5kJ/mol,为正值,表明温度升高,CM值增加,聚合度降低。")
104
由于氯乙烯的CM值较大,聚氯乙烯的分子量可由温度控制,与引发剂的用量基本无关;聚合速率由引发剂浓度控制。这在工艺上是十分方便的。
同样可得到向甲基丙烯酸甲酯的转移常数与温度间的关 系式。 式中1.93 kJ/mol为转移活化能与增长能的差值。可见温 度对甲基丙烯酸甲酯聚合的聚合度影响很小。

105
4、向引发剂转移 向引发剂转移实际上就是引发剂在自由基作用下的诱导分解。由此可见,诱导分解不仅影响引发剂效率,还影响聚合物的分子量。
(1)向引发剂转移常数CI的测定 采用过氧化物作为引发剂的本体聚合存在向引发剂转移,平均聚合度的倒数可表示为: 无向引发剂转移时,聚合度的倒数与Rp为一次方关系, 存在向引发剂转移时,聚合度的倒数与Rp为二次方关系。
向引发剂转移常数CI的测定. 采用过氧化物作为引发剂的本体聚合存在向引发剂转移,平均聚合度的倒数可表示为: 无向引发剂转移时,聚合度的倒数与Rp为一次方关系, 存在向引发剂转移时,聚合度的倒数与Rp为二次方关系。")
106
聚苯乙烯聚合度的倒数和聚合速率的关系 AIBN—偶氮二异丁腈; BPO—过氧化二苯甲酰; CHP—异丙苯过氧化;
t-BHP— 特丁基过氧化氢 聚苯乙烯聚合度的倒数和聚合速率的关系

107
i、引发剂转移常数CI的第一种求法: 以上式左边对Rp作图,可由直线斜率求出CI 。 i i 、引发剂转移常数CI的第二种求法: 以上式左边对[I]/[M]作图,从直线斜率可求出CI,由截距求出CM 。
![i、引发剂转移常数CI的第一种求法: 以上式左边对Rp作图,可由直线斜率求出CI 。 i i 、引发剂转移常数CI的第二种求法: 以上式左边对[I]/[M]作图,从直线斜率可求出CI,由截距求出CM 。](http://slidesplayer.com/slide/11130699/59/images/107/i%E3%80%81%E5%BC%95%E5%8F%91%E5%89%82%E8%BD%AC%E7%A7%BB%E5%B8%B8%E6%95%B0CI%E7%9A%84%E7%AC%AC%E4%B8%80%E7%A7%8D%E6%B1%82%E6%B3%95%EF%BC%9A+%E4%BB%A5%E4%B8%8A%E5%BC%8F%E5%B7%A6%E8%BE%B9%E5%AF%B9Rp%E4%BD%9C%E5%9B%BE%EF%BC%8C%E5%8F%AF%E7%94%B1%E7%9B%B4%E7%BA%BF%E6%96%9C%E7%8E%87%E6%B1%82%E5%87%BACI+%E3%80%82+i+i+%E3%80%81%E5%BC%95%E5%8F%91%E5%89%82%E8%BD%AC%E7%A7%BB%E5%B8%B8%E6%95%B0CI%E7%9A%84%E7%AC%AC%E4%BA%8C%E7%A7%8D%E6%B1%82%E6%B3%95%EF%BC%9A+%E4%BB%A5%E4%B8%8A%E5%BC%8F%E5%B7%A6%E8%BE%B9%E5%AF%B9%5BI%5D%2F%5BM%5D%E4%BD%9C%E5%9B%BE%EF%BC%8C%E4%BB%8E%E7%9B%B4%E7%BA%BF%E6%96%9C%E7%8E%87%E5%8F%AF%E6%B1%82%E5%87%BACI%EF%BC%8C%E7%94%B1%E6%88%AA%E8%B7%9D%E6%B1%82%E5%87%BACM+%E3%80%82.jpg "i、引发剂转移常数CI的第一种求法: 以上式左边对Rp作图,可由直线斜率求出CI 。 i i 、引发剂转移常数CI的第二种求法: 以上式左边对[I]/[M]作图,从直线斜率可求出CI,由截距求出CM 。")
108
(2)常见引发剂的转移常数CI大小比较 i、偶氮类引发剂通常认为不发生链转移反应,但近年来的研究表明,这类引发剂也有较小的CI值,可能通过下列的置换反应发生链转移。 ii、过氧化物中O-O键较弱,容易发生向过氧化物的链转移,尤以酰基过氧化物为易。 iii、氢过氧化物是引发剂中最强的链转移,转移时可能发生夺氢反应。

109
(3)引发剂浓度对聚合度的影响 而 从以上两方程可知:引发剂浓度对聚合度的影响表现在两个方面:一是正常的引发反应,即影响链增长反应速率,第一项;另一是向引发剂转移,第三项。 向引发剂转移引起的分子量下降不如向单体转移明显。

110
5、向溶剂或链转移剂转移 进行溶液聚合时,必须考虑向溶剂转移对分子量的影响。 (1)向溶剂或链转移剂转移常数CS 的测定
当体系只发生向溶剂或链转移剂的链转移反应,或其它形式的链转移常数很小,相比之下可忽略时,则平均聚合度的倒数可表示为: [S] = CS Xn Xn [M] 式中(1/ Xn)0代表无溶剂时的聚合度的倒数。以1/ Xn 对[S]/[M]作图,由斜率可求得向溶剂的链转移常数CS 。
0代表无溶剂时的聚合度的倒数。以1/ Xn 对[S]/[M]作图,由斜率可求得向溶剂的链转移常数CS 。")
111
(2)CS 与自由基种类、溶剂种类和温度等因素有关系
i、自由基种类(或单体):自由基活性越大, CS一般也越大。如苯乙烯的自由基活性较小(共轭效应), CS较小;而醋酸乙烯酯的自由基活性较大,则CS也较大。 i i 、溶剂种类:溶剂的极性和结构影响CS值。 非极性溶剂的链转移活性对各种条件都相同,极性溶剂受单体的极性影响,规律是:溶剂的极性与单体的极性相反时,链转移活性增高,原因是电子给体和电子受体之间发生部分电子转移,使过渡状态稳定。 对于具有比较活泼氢原子或卤原子的溶剂,链转移常数一般较大。
:自由基活性越大, CS一般也越大。如苯乙烯的自由基活性较小(共轭效应), CS较小;而醋酸乙烯酯的自由基活性较大,则CS也较大。 i i 、溶剂种类:溶剂的极性和结构影响CS值。 非极性溶剂的链转移活性对各种条件都相同,极性溶剂受单体的极性影响,规律是:溶剂的极性与单体的极性相反时,链转移活性增高,原因是电子给体和电子受体之间发生部分电子转移,使过渡状态稳定。 对于具有比较活泼氢原子或卤原子的溶剂,链转移常数一般较大。")
112
某些取代芳烃、卤代烃和醇类的CS大小顺序:
异丙苯>乙苯>甲苯>苯(苯环侧基上的氢较苯环上氢易被夺取) RI > RBr > RCl(C-Cl、 C-Br键较弱,易链转移) CCl4 > CHCl3 > CH2Cl2 R2CHOH > RCH2OH > CH3OH i i i 、提高温度一般可使链转移常数增大。 因为CS = ktr,s/kp ,链转移活化能比增长活化能一般要大17~63kJ/mol ,升高温度对ktr,s的增加比kp的增加要大的多,故CS值增大。
 RI > RBr > RCl(C-Cl、 C-Br键较弱,易链转移) CCl4 > CHCl3 > CH2Cl2. R2CHOH > RCH2OH > CH3OH. i i i 、提高温度一般可使链转移常数增大。 因为CS = ktr,s/kp ,链转移活化能比增长活化能一般要大17~63kJ/mol ,升高温度对ktr,s的增加比kp的增加要大的多,故CS值增大。")
113
链转移剂--分子量调节剂 特指链转移常数较大的小分子物质,通常CS为1或更大;可以为溶剂。 在工艺上,有时有意在聚合体系中加入某些链转移常数较大的溶剂来调节、控制分子量。 例如生产丁苯橡胶时加入的硫醇; 生产低分子量聚氯乙烯时加入的三氯乙烯; 生产聚乙烯或聚丙烯时加入的氢气等。 脂肪族硫醇、三氯乙烯、四氯甲烷等多是最常用的分子量调节剂。

114
分子量调节剂一般选用CS≈1的化合物。因此时ktr
分子量调节剂一般选用CS≈1的化合物。因此时ktr.s≈kp,消耗分子量调节剂和消耗单体的速率接近,聚合过程中可保持[S]/[M]大致不变。 CS太小用量太多, CS太大则早期就消耗,对分子量控制不利。 不同链自由基对同一种分子量调节剂的转移常数并不一定相同。如十二硫醇在苯乙烯聚合中的CS为19,在丙烯腈聚合中则为0.73,而在丁二烯/苯乙烯共聚时为0.66 。

115
6、向聚合物的链转移 向聚合物的链转移反应在低转化率时,由于聚合物浓度很小,通常可忽略不计;但当转化率较高,聚合物浓度较大时,需考虑。
由于向聚合物的链转移结果并不一定降低聚合物的平均动力学链长(或聚合度),故在研究向聚合物链转移时,主要是阐明产物的结构,而不是推算链转移常数 。 (1)形成具有长支链的聚合物 向聚合物转移结果,主要是在主链上形成活性点,单体在该活性点上加成增长,形成较长的支链。
,故在研究向聚合物链转移时,主要是阐明产物的结构,而不是推算链转移常数 。 (1)形成具有长支链的聚合物. 向聚合物转移结果,主要是在主链上形成活性点,单体在该活性点上加成增长,形成较长的支链。")
116
(2)形成具有短支链的聚合物 高压聚乙烯中的乙基和丁基短支链的形成是因为链自由基与本身链中的亚甲基上的氢发生了链转移反应。
形成具有短支链的聚合物 高压聚乙烯中的乙基和丁基短支链的形成是因为链自由基与本身链中的亚甲基上的氢发生了链转移反应。")
117
高压聚乙烯分子中的短支链数可高达30个支链/500个单体单元。
聚氯乙烯也是容易发生转移的大分子,每1000个单体单元中约含有10~20个支链。 向大分子转移不影响产物的平均分子量,但使得分子量分布变宽。

118
3.7 阻聚和缓聚 一、阻聚剂和阻聚机理 上节中已经提到,链转移对聚合速率的影响有三种情况:
3.7 阻聚和缓聚 一、阻聚剂和阻聚机理 上节中已经提到,链转移对聚合速率的影响有三种情况: (1)新自由基的活性与原自由基活性相同,聚合速率不变; (2)新自由基活性减弱,出现缓聚现象; (3)新自由基没有引发活性,聚合停止,表现为阻聚作用。 实际上,阻聚和缓聚只是程度上的差别,并与严格的区分界限。
新自由基的活性与原自由基活性相同,聚合速率不变; (2)新自由基活性减弱,出现缓聚现象; (3)新自由基没有引发活性,聚合停止,表现为阻聚作用。 实际上,阻聚和缓聚只是程度上的差别,并与严格的区分界限。")
119
阻聚现象在高分子科学与工业中十分重要。 单体中的杂质可能会阻碍聚合的正常进行,因此必须对单 体进行精制。 单体在加热精制和贮存运输过程中要防止其自聚,需要加 入一定量的阻聚剂。使用时再脱除阻聚剂。 某些单体在聚合时为了得到一定结构或分子量的产物,需 控制转化率。因此在聚合到一定转化率时需加入阻聚剂,使 聚合反应终止。 在高分子化学研究中,利用高效阻聚剂捕捉自由基的能力 测定引发速率。

120
1、阻聚剂和缓聚剂的概念 阻聚剂(inhibitor)-能与链自由基反应生成非自由基或不能引发单体聚合的低活性自由基而使聚合反应完全停止的化合物称为阻聚剂; 缓聚剂(retarding agents)--能使聚合反应速率减慢的化合物称为缓聚剂。 当体系中存在阻聚剂时,在聚合反应开始以后(引发剂开始分解),并不能马上引发单体聚合,必须在体系中的阻聚剂全部消耗完后,聚合反应才会正常进行。即从引发剂开始分解到单体开始转化存在一个时间间隔,称诱导期 (induction period, ti).
,并不能马上引发单体聚合,必须在体系中的阻聚剂全部消耗完后,聚合反应才会正常进行。即从引发剂开始分解到单体开始转化存在一个时间间隔,称诱导期 (induction period, ti).")
121
阻聚剂会导致聚合反应存在诱导期,但在诱导期过后,不会改变聚合速率。
缓聚剂并不会使聚合反应完全停止,不会导致诱导期,只会减慢聚合反应速率。 但有些化合物兼有阻聚作用与缓聚作用,即在一定的反应阶段充当阻聚剂,产生诱导期,反应一段时间后其阻聚作用消失,转而成为缓聚剂,使聚合反应速率减慢。

122
I 无阻聚剂与缓聚剂 II 加阻聚剂 单体转化率 III 加缓聚剂 IV 兼有阻聚与缓聚作用 ti 诱导期 时间
苯乙烯100℃热聚合的阻聚作用

123
2、几类典型的阻聚剂和阻聚机理 分子型阻聚剂 自由基型阻聚剂 加成型阻聚剂 链转移型阻聚剂 电荷转移型阻聚剂 (1)分类 i、按组成结构
苯醌、硝基化合物、芳胺、酚类、含硫化合物等; 1,1-二苯基-2-三硝基苯肼(DPPH)等。 ii、按阻聚剂和自由基反应的机理 加成型阻聚剂 链转移型阻聚剂 电荷转移型阻聚剂
等。 ii、按阻聚剂和自由基反应的机理. 加成型阻聚剂. 链转移型阻聚剂. 电荷转移型阻聚剂.")
124
(2)阻聚剂的阻聚机理 i、加成型阻聚剂 与链自由基快速加成,使之转化为活性低的自由基,从而起到阻聚剂或缓聚剂的作用。
是目前最常用的阻聚剂类型,常见的有氧气、硫、苯醌衍生物和硝基化合物等。 苯醌分子上的氧和碳原子都可与自由基加成,然后通过偶合或歧化终止。

125
苯醌 苯醌的阻聚行为比较复杂,可能涉及下列一些加成和氢转移反应:
上述形成的取代苯醌还可继续进行阻聚反应,再消灭一个自由基。因此每一个苯醌分子能终止2个自由基。

126
对苯二酚价格便宜,氧又是空气中存在的,取之十分方便。因此对苯二酚不失为一种经济实用的阻聚剂,在工业上和实验室中广泛使用。
对苯二酚本身的阻聚能力不强,但在氧的存在下容易氧化成苯醌,从而提高了阻聚能力。 对苯二酚价格便宜,氧又是空气中存在的,取之十分方便。因此对苯二酚不失为一种经济实用的阻聚剂,在工业上和实验室中广泛使用。

127
芳族硝基化合物 也是一种常用的加成型阻聚剂,其阻聚机理可能为: 自由基向苯环或硝基进攻。 硝基化合物对比较活泼的富电自由基的阻聚效果好。
随苯环上硝基数增多,芳族硝基化合物的阻聚效率增加。

128
氧 氧的阻聚行为比较复杂。在低温下,氧是很好的阻聚剂。因此聚合反应一般要在去除氧的情况下进行。
聚合物过氧化物在低温下很稳定,但在高温时却可分解成活性很大的自由基,可引发聚合。因此氧在高温时是很好的引发剂。例如乙烯的高温高压聚合(高压聚乙烯)就是以氧为引发剂的。
就是以氧为引发剂的。")
129
DPPH ii、链转移型阻聚剂 主要有1,1-二苯基-2-三硝基苯肼(DPPH)、芳胺、酚类等。
是自由基型阻聚剂,效率极高,浓度为10-4 mol/L就 足以使单体阻聚。 DPPH分子能够化学计量地消灭一个自由基,素有自由基扑捉剂之称。 DHHP为黑色,捕捉自由基后变为无色,因此可通过比色法测定引发速率。

130
仲胺也是通过转移反应实现阻聚作用的。 苯胺和苯酚的阻聚效率很低,既使对十分活泼的醋酸乙 烯酯也仅是效果很差的缓聚剂。但苯环上有多个供电的烷基 取代后,缓聚效果可显著增加。对苯二酚经氧化后转变成苯 醌,阻聚效果大大增加。

131
iii、电荷转移型阻聚剂及其机理 电荷转移型阻聚剂的典型代表是氯化铁和氯化铜。其阻聚机理如下: 氯化铁和氯化铜的阻聚效率很高,能1 对 1按化学计量消灭自由基,因此可用于测定引发速率。 工业上应避免使用碳钢或铜质的反应釜和管道,以防阻聚发生。

132
二、烯丙基单体的自阻聚作用 烯丙基单体(CH2=CH—CH2Y)聚合往往只能形成低聚物,这时因为烯丙基单体有自阻聚作用。
聚合往往只能形成低聚物,这时因为烯丙基单体有自阻聚作用。")
133
发单体,而只能与初级自由基或自身进行双基终止,因此表 现为自阻聚作用。
所形成的烯丙基自由基有高度的共振稳定性,不能再引 发单体,而只能与初级自由基或自身进行双基终止,因此表 现为自阻聚作用。 醋酸烯丙酯(CH2=CH—CH2OCOCH3)是典型的烯丙基 单体,聚合速率很低,聚合度只能达14。与引发剂浓度呈一 级反应。这些都是衰减链转移的结果。
是典型的烯丙基. 单体,聚合速率很低,聚合度只能达14。与引发剂浓度呈一. 级反应。这些都是衰减链转移的结果。")
134
烯丙基单体聚合低活性的原因: 单体活性不高且加成反应生成的链自由基是二级碳自由基,不稳定,不利于加成反应的进行;
由于烯丙基氢很活泼,且链转移后生成的烯丙基自由基由于有双键的共振作用非常稳定,因此对链转移反应非常有利。这样,由于链转移反应极易发生,ktr>>kp,烯丙基单体聚合只能得到低聚物, 由于链转移生成的烯丙基自由基很稳定。不能引发单体聚合,只能与其它自由基终止,起缓聚或阻聚作用。

135
丙烯、异丁烯都属于烯丙基单体,对自由基聚合的活性很
低,只能进行配位聚合(丙烯)和阳离子聚合(异丁烯)。 丁二烯也是一种烯丙基单体,其自由基十分稳定。但丁二 烯单体十分活泼,因此尚能进行均聚反应。但对氯乙烯、醋 酸乙烯酯等不活泼单体却是阻聚剂。 甲基丙烯酸甲酯、甲基丙烯腈等单体虽然也都是烯丙基单 体,但因酯基和腈基对自由基有稳定作用,降低了自由基的 链转移活性,同时单体又较活泼,因此链转移衰减不明显, 仍可聚合得到高分子。
和阳离子聚合(异丁烯)。 丁二烯也是一种烯丙基单体,其自由基十分稳定。但丁二. 烯单体十分活泼,因此尚能进行均聚反应。但对氯乙烯、醋. 酸乙烯酯等不活泼单体却是阻聚剂。 甲基丙烯酸甲酯、甲基丙烯腈等单体虽然也都是烯丙基单. 体,但因酯基和腈基对自由基有稳定作用,降低了自由基的. 链转移活性,同时单体又较活泼,因此链转移衰减不明显, 仍可聚合得到高分子。")
136
3.8 聚合热力学 一、聚合热力学的一般概念 一种单体能否反应成为聚合物,可从其聚合前后的自由能变化来判断。
对于聚合过程,单体是初态,自由能为G1,聚合物为终态,自由能为G2。当ΔG = G2-G1<0 时,聚合过程可自发进行; ΔG >0 ,聚合物解聚成单体; ΔG = 0,单体与聚合物处于平衡状态。 自由能 ΔG 与聚合反应焓变ΔH和熵变ΔS的关系为:

137
单体转化为聚合物,无序性减小,熵值减小,因此ΔS总
是负值。一般为-105~-125 J/mol.K。在通常聚合温度下 (室温~100℃),T ΔS= -30~-45 J/mol。因此要使聚合 体系的ΔG <0 , ΔH须为负值(放热),数值上必须超过 45 J/mol。 聚合热 烯类单体的聚合热可由键能作大概的估算。
,T ΔS= -30~-45 J/mol。因此要使聚合. 体系的ΔG <0 , ΔH须为负值(放热),数值上必须超过. 45 J/mol。 聚合热. 烯类单体的聚合热可由键能作大概的估算。")
138
聚合的结果是一个双键转变为两个单键。C—C单键的键能约为350 kJ/mol,双键的键能约610 kJ/mol。因此无取代基时,烯类单体的聚合热约为-90 kJ/mol。
单纯双键的聚合从热力学上看是可行的。实际上,大多数烯类单体的聚合热低于估算值,原因是存在取代基的位阻效应、共轭效应、氢键作用、溶剂化作用等。 根据热力学方程,ΔH = ΔE-p ΔV。聚合反应为等容过程,ΔV=0,则ΔH = ΔE。即聚合热等于内能的变化,这是聚合热受取代基影响的内在因素。

139
(1)位阻效应 位阻效应使聚合热降低。如乙烯ΔH = -95 kJ/mol,双取代后的异丁烯(-51.5 kJ/mol),MMA(-56.5kJ/mol),α-甲基苯乙烯( -35.1 kJ/mol),都比乙烯低得多。但取代的单体聚合热下降不多,如丙烯(-85.8kJ/mol)、1-丁烯 ( -79.5kJ/mol)。 甲醛的ΔH = -50.23 kJ/mol,引入甲基变成乙醛后,聚合热降至0 kJ/mol,常温下不可能聚合。
。 甲醛的ΔH = -50.23 kJ/mol,引入甲基变成乙醛后,聚合热降至0 kJ/mol,常温下不可能聚合。")
140
(2)共轭效应 共轭效应也使聚合热降低。苯乙烯(-69.9kJ/mol)、丁二烯(-72.8kJ/mol)、异戊二烯(-74.5kJ/mol)、丙烯腈(-72.4kJ/mol)都是共轭单体,因此聚合热都降低。 丙烯酸(-66.9kJ/mol)、丙烯酸甲酯(-78.7kJ/mol)也有一定共轭效应,聚合热也有所降低。 丙烯(-85.8kJ/mol)分子上的甲基有超共轭效应,聚合热比乙烯略低。α-甲基苯乙烯( -35.1 kJ/mol)既有苯基的共轭,又有甲基的超共轭,而且有双取代的位阻效应,因此聚合热降低很多。
、丙烯酸甲酯(-78.7kJ/mol)也有一定共轭效应,聚合热也有所降低。 丙烯(-85.8kJ/mol)分子上的甲基有超共轭效应,聚合热比乙烯略低。α-甲基苯乙烯( -35.1 kJ/mol)既有苯基的共轭,又有甲基的超共轭,而且有双取代的位阻效应,因此聚合热降低很多。")
141
(3)取代基的电负性 取代基的电负性大,有利于聚合热的升高。例如氯乙烯(-95.8kJ/mol)、硝基乙烯(-90.8kJ/mol)、偏二氟乙烯(-129.7kJ/mol)、四氟乙烯(-154.8kJ/mol)。 偏二氯乙烯(-75.3kJ/mol)为双取代单体,但氯原子电负性大,两者抵消,聚合热降低不多。 含氟聚合物的聚合热特别高,可能与这类单体的C—C键单键能较大有关。如六氟乙烷中C—C键能为520 kJ/mol,而乙烷中C—C键能为350 kJ/mol。
为双取代单体,但氯原子电负性大,两者抵消,聚合热降低不多。 含氟聚合物的聚合热特别高,可能与这类单体的C—C键单键能较大有关。如六氟乙烷中C—C键能为520 kJ/mol,而乙烷中C—C键能为350 kJ/mol。")
142
(4)氢键和溶剂化作用 氢键和溶剂化作用都使得聚合物分子运动受阻,内能增加,因此聚合热降低。 如氢键的影响:丙烯酸(-66.9kJ/mol)、甲基丙烯酸(-42.3kJ/mol,氢键+双取代)。溶剂化作用:丙烯酰胺(纯-82.0kJ/mol,苯中-60.2kJ/mol)、甲基丙烯酰胺(苯中-35.1kJ/mol,溶剂化+双取代)。
、甲基丙烯酸(-42.3kJ/mol,氢键+双取代)。溶剂化作用:丙烯酰胺(纯-82.0kJ/mol,苯中-60.2kJ/mol)、甲基丙烯酰胺(苯中-35.1kJ/mol,溶剂化+双取代)。")
143
理论上,任何聚合反应都存在逆反应。但因多数单体聚
二、聚合上限温度 1、 聚合上限温度的概念 理论上,任何聚合反应都存在逆反应。但因多数单体聚 合热较大,在一般聚合温度下逆反应速率很低,可忽略。而 在高温下,TΔS变得很大,逆反应不可忽略。如PMMA在 高温下可分解出单体。 当单体与聚合物处于平衡状态时,ΔG = 0,即

144
因此: Tc称为聚合上限温度。超过这一温度,聚合无法进行。 聚合上限温度与单体浓度的关系 链增长反应和解聚反应为一对竞争反应。

145
两者的速率方程为: 平衡时: 根据自由基等活性原理, 可得聚合平衡常数:

146
在热力学非标准状态下,化学反应的自由能变化方程为:
其中ΔG0为标准状态下( Pa,活度为1)的自由能变 化。平衡时,ΔG = 0,因此有: 则:
的自由能变. 化。平衡时,ΔG = 0,因此有: 则:")
147
从式2-10-10可见,体系中单体浓度不同时,Tc是不同的。因此有一系列不确定的上限温度。在学术研究中规定:平衡浓度[M]e=1 mol/L 时的平衡温度称为聚合上限温度。此时,
热力学概念十分明确。 在工程上规定:聚合物浓度为0(单体浓度为100%)时的温度为聚合上限温度。求取方便,可将转化率—温度曲线外推到0,此时温度即为Tc。
![从式 可见,体系中单体浓度不同时,Tc是不同的。因此有一系列不确定的上限温度。在学术研究中规定:平衡浓度[M]e=1 mol/L 时的平衡温度称为聚合上限温度。此时,](http://slidesplayer.com/slide/11130699/59/images/147/%E4%BB%8E%E5%BC%8F+%E5%8F%AF%E8%A7%81%EF%BC%8C%E4%BD%93%E7%B3%BB%E4%B8%AD%E5%8D%95%E4%BD%93%E6%B5%93%E5%BA%A6%E4%B8%8D%E5%90%8C%E6%97%B6%EF%BC%8CTc%E6%98%AF%E4%B8%8D%E5%90%8C%E7%9A%84%E3%80%82%E5%9B%A0%E6%AD%A4%E6%9C%89%E4%B8%80%E7%B3%BB%E5%88%97%E4%B8%8D%E7%A1%AE%E5%AE%9A%E7%9A%84%E4%B8%8A%E9%99%90%E6%B8%A9%E5%BA%A6%E3%80%82%E5%9C%A8%E5%AD%A6%E6%9C%AF%E7%A0%94%E7%A9%B6%E4%B8%AD%E8%A7%84%E5%AE%9A%EF%BC%9A%E5%B9%B3%E8%A1%A1%E6%B5%93%E5%BA%A6%5BM%5De%3D1+mol%2FL+%E6%97%B6%E7%9A%84%E5%B9%B3%E8%A1%A1%E6%B8%A9%E5%BA%A6%E7%A7%B0%E4%B8%BA%E8%81%9A%E5%90%88%E4%B8%8A%E9%99%90%E6%B8%A9%E5%BA%A6%E3%80%82%E6%AD%A4%E6%97%B6%EF%BC%8C.jpg "热力学概念十分明确。 在工程上规定:聚合物浓度为0(单体浓度为100%)时的温度为聚合上限温度。求取方便,可将转化率—温度曲线外推到0,此时温度即为Tc。")
148
在聚合上限温度以上,单体难以聚合,任何努力都是徒劳的。
但是在聚合物形成以后,某些聚合物可在聚合上限温度以上使用。这是因为聚合物的解聚需要时间和解聚中心,因此是处于假平衡状态。在适当条件和适当时间内,解聚仍会发生。如处于高温时间足够长,体系中存在残留引发剂、残留单体等,都可作为解聚中心而使解聚发生。

149
2、影响聚合上限温度的因素 (1)ΔH和ΔS的影响 影响ΔH和ΔS的因素,均影响Tc。如聚合物中单体单元间的键合力越大,越不易降解,则Tc越高。侧基的空间张力和空间位阻越大,则Tc越低。单体的共振稳定性使得ΔH降低,故Tc也降低。

150
压力对Tc有很大影响。聚合时体积收缩的过程。增加 压力有利于平衡向形成聚合物的方向移动进行。
(2)压力的影响 压力对Tc有很大影响。聚合时体积收缩的过程。增加 压力有利于平衡向形成聚合物的方向移动进行。 压力对Tc的影响可从Clapeyron—Clausius方程解释。 由于ΔV和ΔH均为负值,则ΔV/ΔH为正值,即Tc随压力上升而提高。 ( )
压力的影响. 压力对Tc有很大影响。聚合时体积收缩的过程。增加 压力有利于平衡向形成聚合物的方向移动进行。 压力对Tc的影响可从Clapeyron—Clausius方程解释。 由于ΔV和ΔH均为负值,则ΔV/ΔH为正值,即Tc随压力上升而提高。 ( )")
151
压力对Tc的影响还可从压力-[M]e关系来解释。由平衡常数可得出以下关系式:
( ) 式中ΔV为负值,故压力上升,[M]e下降,则Tc上升。 对烯类单体而言,有利于聚合的条件是高压低温 。
![压力对Tc的影响还可从压力-[M]e关系来解释。由平衡常数可得出以下关系式:](http://slidesplayer.com/slide/11130699/59/images/151/%E5%8E%8B%E5%8A%9B%E5%AF%B9Tc%E7%9A%84%E5%BD%B1%E5%93%8D%E8%BF%98%E5%8F%AF%E4%BB%8E%E5%8E%8B%E5%8A%9B%EF%BC%8D%5BM%5De%E5%85%B3%E7%B3%BB%E6%9D%A5%E8%A7%A3%E9%87%8A%E3%80%82%E7%94%B1%E5%B9%B3%E8%A1%A1%E5%B8%B8%E6%95%B0%E5%8F%AF%E5%BE%97%E5%87%BA%E4%BB%A5%E4%B8%8B%E5%85%B3%E7%B3%BB%E5%BC%8F%EF%BC%9A.jpg "( ) 式中ΔV为负值,故压力上升,[M]e下降,则Tc上升。 对烯类单体而言,有利于聚合的条件是高压低温 。")
Similar presentations













.>")
 中国地质大学(北京)材料科学与工程学院.>")



 常数项级数的概念 袁安锋 2016.7.>")
