Download presentation
Presentation is loading. Please wait.

1
第四章 高聚物的结构 第一节高聚物的合成 1.1加聚反应 反应活性中心----- 自由基聚合; 离子聚合 阳离子聚合 阴离子聚合
烯类单体通过双键打开发生的加成聚合反应。 反应活性中心 自由基聚合; 离子聚合 大多属于连锁聚合。 连锁聚合反应通常由链引发、链增长和链终止等基元反 应组成。每一步的速度和活化能相差很大。 阳离子聚合 阴离子聚合 配位聚合

2
聚合过程中有时还会发生链转移反应,但不是必须经过的基元反应。

3
均裂的结果产生两个自由基;异裂的结果形成阴离子和 阳离子。
引发剂分解成活性中心时,共价键有两种裂解形式:均 裂和异裂。 均裂的结果产生两个自由基;异裂的结果形成阴离子和 阳离子。 自由基、阴离子和阳离子均有可能作为连锁聚合的活性 中心,因此有自由基聚合、阴离子聚合和阳离子聚合之分。

4
1.2 连锁聚合的单体 和环状化合物。 不同单体对聚合机理的选择性受共价键断裂后的电子结 构控制。
连锁聚合的单体包括单烯类、共轭二烯类、炔类、羰基 和环状化合物。 不同单体对聚合机理的选择性受共价键断裂后的电子结 构控制。 醛、酮中羰基双键上C和O的电负性差别较大,断裂后具 有离子的特性,因此只能由阴离子或阳离子引发聚合,不能 进行自由基聚合。环状单体一般也按阴离子或阳离子机理进 行聚合。

5
烯类单体的碳—碳双键既可均裂,也可异裂,因此可进
行自由基聚合或阴、阳离子聚合,取决于取代基的诱导效应 和共轭效应。 乙烯分子中无取代基,结构对称,因此无诱导效应和共 轭效应。只能在高温高压下进行自由基聚合,得到低密度聚 乙烯。在配位聚合引发体系引发下也可进行常温低压配位聚 合,得到高密度聚乙烯。

6
基等,碳—碳双键上电子云增加,有利于阳离子聚合进行。
分子中含有推电子基团,如烷基、烷氧基、苯基、乙烯 基等,碳—碳双键上电子云增加,有利于阳离子聚合进行。 丙烯分子上有一个甲基,具有推电子性和超共轭双重效 应,但都较弱,不足以引起阳离子聚合,也不能进行自由基 聚合。只能在配位聚合引发体系引发下进行配位聚合。 其他含有一个烷基的乙烯基单体也具有类似的情况。

7
结论: 1,1取代的异丁烯分子中含有两个甲基,推电子能力大大 增强,可进行阳离子聚合,但不能进行自由基聚合。
含有烷氧基的烷氧基乙烯基醚、苯基的苯乙烯、乙烯基 的丁二烯均可进行阳离子聚合。 结论: 含有1,1-双烷基、烷氧基、苯基和乙烯基的烯烃因推电 子能力较强,可进行阳离子聚合。

8
酯)等,碳—碳双键上电子云密度降低,并使形成的阴离子 活性种具有共轭稳定作用,因此有利于阴离子聚合进行。
分子中含有吸电子基团,如:腈基、羰基(醛、酮、酸、 酯)等,碳—碳双键上电子云密度降低,并使形成的阴离子 活性种具有共轭稳定作用,因此有利于阴离子聚合进行。 例如丙烯腈中的腈基能使负电荷在碳—氮两个原子上离 域共振而稳定。
等,碳—碳双键上电子云密度降低,并使形成的阴离子. 活性种具有共轭稳定作用,因此有利于阴离子聚合进行。 例如丙烯腈中的腈基能使负电荷在碳—氮两个原子上离. 域共振而稳定。")
9
卤素原子既有诱导效应(吸电子),又有共轭效应(推
电子),但两者均较弱,因此既不能进行阴离子聚合,也不 能进行阳离子聚合,只能进行自由基聚合。如氯乙烯、氟乙 烯、四氟乙烯均只能按自由基聚合机理进行。 除了少数含有很强吸电子基团的单体(如偏二腈乙烯、 硝基乙烯)只能进行阴离子聚合外,大部分含吸电子基团的 单体均可进行自由基聚合。 含有共轭双键的烯类单体,如苯乙烯、α-苯乙烯、丁二 烯、异戊二烯等,因电子云流动性大,容易诱导极化,因此 既可进行自由基聚合,也可进行阴、阳离子聚合。
,但两者均较弱,因此既不能进行阴离子聚合,也不. 能进行阳离子聚合,只能进行自由基聚合。如氯乙烯、氟乙. 烯、四氟乙烯均只能按自由基聚合机理进行。 除了少数含有很强吸电子基团的单体(如偏二腈乙烯、 硝基乙烯)只能进行阴离子聚合外,大部分含吸电子基团的. 单体均可进行自由基聚合。 含有共轭双键的烯类单体,如苯乙烯、α-苯乙烯、丁二. 烯、异戊二烯等,因电子云流动性大,容易诱导极化,因此. 既可进行自由基聚合,也可进行阴、阳离子聚合。")
10
结论: 合的选择性很小,大部分烯类单体均可进行自由基聚合。 取代基对乙烯基单体聚合机理的影响如下:
乙烯基单体对离子聚合有较强的选择性,但对自由基聚 合的选择性很小,大部分烯类单体均可进行自由基聚合。 取代基对乙烯基单体聚合机理的影响如下:

11
表4—1 常见烯类单体的聚合类型 续表 单体 聚合类型 中文名称 分子式 自由基 阴离子 阳离子 配位 氟乙烯 CH2=CHF ⊕ 四氟乙烯
表4—1 常见烯类单体的聚合类型 续表 单体 聚合类型 中文名称 分子式 自由基 阴离子 阳离子 配位 氟乙烯 CH2=CHF ⊕ 四氟乙烯 CF2=CF2 六氟丙烯 CF2=CFCF3 偏二氟乙烯 CH2=CF2 烷基乙烯基醚 CH2=CH—OR 醋酸乙烯酯 CH2=CHOCOCH3 丙烯酸甲酯 CH2=CHCOOCH3 + 甲基丙烯酸甲酯 CH=C(CH3)COOCH3 丙烯腈 CH2=CHCN 偏二腈乙烯 CH2=C(CN)2 硝基乙烯 CH2=CHNO2
COOCH3. 丙烯腈. CH2=CHCN. 偏二腈乙烯. CH2=C(CN)2. 硝基乙烯. CH2=CHNO2.")
12
由取代基的体积、数量和位置等因素所引起的空间位阻
作用,对单体的聚合能力有显著影响,但不影响其对活性种 的选择性。 单取代烯类单体, 即使取代基体积较大,也不妨碍聚合, 如乙烯基咔唑。 1,1双取代的烯类单体,因分子结构对称性更差,极化程 度增加,因此更容易聚合。取代基体积较大时例外,如1,1- 二苯乙烯不能聚合。

13
不论氟代的数量和位置,均极易聚合。 1,2双取代的烯类化合物,因结构对称,极化程度低,位
阻效应大,一般不能聚合。但有时能与其他单体共聚,如马 来酸酐能与苯乙烯共聚。 三取代、四取代的烯类化合物一般不能聚合,但氟代乙 烯例外。例如:氟乙烯、1,1-二氟乙烯、1,2-二氟乙烯、三氟 乙烯、四氟乙烯均可聚合。 不论氟代的数量和位置,均极易聚合。 原因: 氟原子半径较小,仅大于氢原子,不会造成空间位阻。

14
表4—2 乙烯基单体取代基的体积与数量对聚合特性的影响
表4—2 乙烯基单体取代基的体积与数量对聚合特性的影响 取代基X 取代基半径 /nm 一取代 二取代 三取代 四取代 1,1-取代 1,2-取代 H 0.032 + F 0.064 Cl 0.099 - CH3 0.109 Br 0.114 I 0.133 C6H5 0.232 * 碳原子半径:0.075nm

15
1.3 自由基聚合机理 考察自由基聚合有两个重要指标:聚合速率和分子量。 为了弄清楚这两个指标的影响因素和控制方法,就必须从自
由基聚合的机理入手。 1.3.1 自由基聚合的基元反应 1)链引发反应 形成单体自由基活性种的反应。引发剂、光能、热能、 辐射能等均能使单体生成单体自由基。
链引发反应. 形成单体自由基活性种的反应。引发剂、光能、热能、 辐射能等均能使单体生成单体自由基。")
16
引发剂分解(均裂)形成自由基,为吸热反应, 活化能 高,反应速度慢。 E = 105~150 kJ/mol (4—1)
由引发剂引发时,由两步反应组成: 初级自由基的生成 引发剂分解(均裂)形成自由基,为吸热反应, 活化能 高,反应速度慢。 E = 105~150 kJ/mol (4—1) kd = 10-4~10-6 s (4—2)
形成自由基,为吸热反应, 活化能. 高,反应速度慢。 E = 105~150 kJ/mol (4—1) kd = 10-4~10-6 s-1 (4—2)")
17
b. 单体自由基的形成 由初级自由基与单体加成产生,为放热反应, 活化能低, 反应速度快。 E = 20 ~ 34 kJ/mol (4—3) 链引发包含第二步,因为这一步反应与后继的链增长反应 相似,有一些副反应可以使某些初级自由基不参与单体自由基 的形成,也就无法链增长。

18
2)链增长反应 链引发反应产生的单体自由基具有继续打开其它单体π键的能力,形成新的链自由基,如此反复的过程即为链增长反应。 两个基本特征: (1)放热反应,聚合热约55 ~ 95kJ/mol。
链增长反应 链引发反应产生的单体自由基具有继续打开其它单体π键的能力,形成新的链自由基,如此反复的过程即为链增长反应。 两个基本特征: (1)放热反应,聚合热约55 ~ 95kJ/mol。")
19
因此,在自由基聚合反应体系内,往往只存在单体和聚合物两部分,不存在聚合度递增的一系列中间产物。
(2)链增长反应活化能低,约为20 ~ 34 kJ/mol,反应速率极高,在0.01 ~几秒钟内聚合度就可达几千至几万,难以控制。 因此,在自由基聚合反应体系内,往往只存在单体和聚合物两部分,不存在聚合度递增的一系列中间产物。
链增长反应活化能低,约为20 ~ 34 kJ/mol,反应速率极高,在0.01 ~几秒钟内聚合度就可达几千至几万,难以控制。 因此,在自由基聚合反应体系内,往往只存在单体和聚合物两部分,不存在聚合度递增的一系列中间产物。")
20
自由基聚合反应中,结构单元间的连接存在“头—尾”、
“头—头”(或“尾—尾”)两种可能的形式,一般以头-尾结 构为主。 原因: (1)头尾连接时,自由基上的独电子与取代基构成共轭体 系,使自由基稳定。而头头连接时无共轭效应,自由基不稳 定。两者活化能相差34 ~ 42 kJ/mol。共轭稳定性较差的单 体,容易出现头头结构。聚合温度升高,头头结构增多。
两种可能的形式,一般以头-尾结. 构为主。 原因: (1)头尾连接时,自由基上的独电子与取代基构成共轭体. 系,使自由基稳定。而头头连接时无共轭效应,自由基不稳. 定。两者活化能相差34 ~ 42 kJ/mol。共轭稳定性较差的单. 体,容易出现头头结构。聚合温度升高,头头结构增多。")
21
(2)以头—尾方式结合时,空间位阻要比头—头方式结合
时的小,故有利于头尾结合。 虽然电子效应和空间位阻效应都有利于生成头尾结构聚 合物,但还不能做到序列结构上的绝对规整。从立体结构来 看,自由基聚合物分子链上取代基在空间的排布是无规的, 因此聚合物往往是无定型的。

22
3)链终止反应 链自由基失去活性形成稳定聚合物的反应。可以分为偶 合终止和歧化终止。 偶合终止:两个链自由基头部的独电子相互结合成共价 键,生成饱和高分子的反应。生成的高分子两端都有引发剂 碎片,聚合度为链自由基重复单元数的两倍。

23
他原子而相互终止的反应。此时生成的高分子只有一端为引 发剂碎片,另一端为饱和或不饱和结构,两者各半,聚合度 与链自由基中的单元数相同。
歧化终止:链自由基夺取另一个自由基上的氢原子或其 他原子而相互终止的反应。此时生成的高分子只有一端为引 发剂碎片,另一端为饱和或不饱和结构,两者各半,聚合度 与链自由基中的单元数相同。

24
偶合终止的活化能约为0,歧化终止的活化能为8 ~
21 kJ/mol。 终止方式与单体种类和聚合条件有关。一般而言,单体位 阻大,聚合温度高,难以偶合终止,多以歧化终止为主。 例如: 60℃以下苯乙烯聚合以几乎全为偶合终止, 60℃以上歧化终止逐步增多。 60℃以下甲基丙烯酸甲酯聚合两种终止方式均有, 60℃以上则以歧化终止逐步为主。

25
4)链转移反应 链自由基从单体、溶剂、引发剂、大分子上夺取原子而 终止,而失去原子的分子成为自由基继续新的增长,使聚合 反应继续进行的过程,称为“链转移反应”。 向低分子转移的结果是使聚合物相对分子质量降低。

26
链转移反应不是自由基聚合必须经过的基元反 应,但具有十分重要的意义。
链自由基可从已形成的大分子上夺取原子而转移,结果 是形成支链型大分子。 链转移反应不是自由基聚合必须经过的基元反 应,但具有十分重要的意义。

27
子化学领域中十分重要。 链自由基向某些物质转移后,所形成的新自由基活性很 低,不足以再引发单体聚合,只能与其他自由基发生双基终
止,导致聚合过程停止。这种现象称为“阻聚反应”。具有阻 聚作用的物质称为“阻聚剂”。如:苯醌、1,1-二苯基-2-三硝 基苯肼(DPPH)等。 阻聚反应不是自由基聚合的基元反应,但在高分 子化学领域中十分重要。
等。 阻聚反应不是自由基聚合的基元反应,但在高分. 子化学领域中十分重要。")
28
1.3.2 自由基聚合反应的特征 (1)可分为链引发、链增长、链终止等基元反应。各基元 反应活化能相差很大。其中链引发反应速率最小,是控制聚 合过程的关键。 慢引发、快增长、速终止。 (2)只有链增长反应使聚合度增加。从单体转化为大分子 的时间极短,瞬间完成。不存在聚合度递增的中间状态(图 2—1)。聚合度与聚合时间基本无关。
。聚合度与聚合时间基本无关。")
29
(3)单体浓度随聚合时间逐步降低,聚合物浓度逐步提高
(图2—2) 。延长聚合时间是为了提高单体转化率。 (4)少量阻聚剂(0.01~0.1%)足以使自由基聚合终止。 图2—1 自由基聚合中分子量与时间的关系 图2—2 自由基聚合中浓度与时间的关系
 。延长聚合时间是为了提高单体转化率。 (4)少量阻聚剂(0.01~0.1%)足以使自由基聚合终止。 图2—1 自由基聚合中分子量与时间的关系. 图2—2 自由基聚合中浓度与时间的关系.")
30
2 缩聚反应 2.1 缩合反应 在有机化学中,典型的缩合反应如醋酸和乙醇的酯化 反应。除了得到主产物醋酸乙酯外,还有副产物水。
2 缩聚反应 2.1 缩合反应 在有机化学中,典型的缩合反应如醋酸和乙醇的酯化 反应。除了得到主产物醋酸乙酯外,还有副产物水。 反应物分子中能参与反应的官能团数称为官能度。醋 酸和乙醇中都只有一个能参与反应的官能团,因此都是单 官能团物质。上述体系称为1—1官能度体系。

31
只要反应体系中有一种原料是单官能度 物质,无论其他原料的官能度为多少,都只 能得到低分子产物。
单官能度的丁醇和二官能度的邻苯二甲酸酐进行酯化反 应,产物为低分子邻苯二甲酸二丁酯,副产物为水。 单官能度的醋酸与三官能度的甘油进行酯化反应,产物 为低分子的三醋酸甘油酯,副产物为水。 只要反应体系中有一种原料是单官能度 物质,无论其他原料的官能度为多少,都只 能得到低分子产物。

32
2.2 缩聚反应 若参与反应的物质均为二官能度的,则缩合反应转化 为缩聚反应。 以二元羧酸与二元醇的聚合反应为例。当一分子二元
酸与一分子二元醇反应时,形成一端为羟基,一端为羧基 的二聚物;二聚物可再与二元酸或二元醇反应,得到两端 均为羟基或均为羧基的三聚体,也可与二聚体反应,得到 四聚体;三聚体既可与单体反应,也可与二聚体或另一种 三聚体反应,如此不断进行,得到高分子量的聚酯。

33
。。。 。。。

34
链增长停止 1.体系粘度增大,低分子不出去 2.原料配比非当量比 3.改变反应条件 如反应温度 反应程度和平衡条件是影响线形缩聚物聚合度的重要因素,但不能用作控制分子量的手段 因为缩聚物的分子两端仍保留着可继续反应的官能团 控制方法:端基封锁 在两官能团等当量的基础上

35
—NH2、—COX(酰卤)、—COOR(酯基)、 —OCOCO—(酸酐)、—H、—X、—SO3H、 —SO2Cl等。
例: 对苯二甲酸与乙二醇反应得到涤纶树脂; 己二胺与己二酸反应得到聚酰胺—6,6; 双酚A与光气反应得到聚碳酸酯; 氨基酸自身聚合得到聚酰胺。 缩聚反应常用的官能团:—OH、—COOH、 —NH2、—COX(酰卤)、—COOR(酯基)、 —OCOCO—(酸酐)、—H、—X、—SO3H、 —SO2Cl等。
、—COOR(酯基)、 —OCOCO—(酸酐)、—H、—X、—SO3H、 —SO2Cl等。")
36
聚合体系中任何两分子(单体或聚合物分子)
基本特征: (1)聚合反应是通过单体官能基之间的反应逐步进行的; (2)每步反应的机理相同,因而反应速率和活化能相同; (3)反应体系始终由单体和分子量递增的一系列中间产物 组成,单体及任何中间产物两分子间都能发生反应; (4)聚合产物的分子量是逐步增大的, (5)反应中有小分子脱出。 聚合体系中任何两分子(单体或聚合物分子) 间都能相互反应生成聚合度更高的聚合物分子。
聚合反应是通过单体官能基之间的反应逐步进行的; (2)每步反应的机理相同,因而反应速率和活化能相同; (3)反应体系始终由单体和分子量递增的一系列中间产物. 组成,单体及任何中间产物两分子间都能发生反应; (4)聚合产物的分子量是逐步增大的, (5)反应中有小分子脱出。 聚合体系中任何两分子(单体或聚合物分子) 间都能相互反应生成聚合度更高的聚合物分子。")
37
2—2官能度体系聚合得到线型聚合物; 2—f(f>2)官能度体系聚合得到支链型 或体型聚合物。
官能度体系聚合得到支链型 或体型聚合物。")
38
缩聚反应的单体转化率、产物聚合度与反应时间关系
示意图: 产物聚合度 单体转化率 反应时间

39
缩聚是可逆反应 根据平衡常数的大小,可将缩聚反应分为三类: ① 平衡常数很小,如聚酯化反应,K≈4,低分子副产物对 分子量有很大影响; ② 平衡常数中等,如聚酰胺化反应,K≈300~400,低分 子副产物对分子量有一定影响; ③ 平衡常数很大,K>1000,实际上可看作不可逆反应,如光气法制备聚碳酸酯。 逐步特性是所有缩聚反应共有的,可逆平衡的程度则各类缩聚反应有明显差别。

40
卡罗瑟斯小传(Wallace Hume Carothers) (1896~1937)
1896年4月27日生于艾奥瓦州伯灵顿, 1920年在密苏里的塔基欧学院毕业; 1921年在伊利诺伊大学获硕士学位; 1924年在伊利诺伊大学获有机化学博 士学位。在该校任教两年后到哈佛大 学任教。 1928年起,在美国杜邦公司任职9年, 领导基础有机化学的研究工作。 1936年当选为美国科学院院士。 1937年4月29日在美国费城一家饭店的 房间里饮用了掺有氰化钾的柠檬汁而 自杀身亡。 主要成果: 1. 合成出氯丁二烯及其聚合物。 2. 以己二酸与己二胺为原料制得 尼龙—66。 一生中发表过60多篇论文和取得近70项专利。

41
弗洛里小传(Paul J. Flory) (1910-1985)
1910年6月19日生于伊利诺伊州斯特灵; 1934年在俄亥俄州州立大学获物理化学 博士学位,后任职于杜邦公司,进行高 分子基础理论研究; 1948年在康奈尔大学任教授; 1953年当选为美国科学院院士; 1957年任梅隆科学研究所执行所长; 1961年任斯坦福大学化学系教授; 1974年获诺贝尔化学奖。 1975年退休; 1985年9月9日逝世。 在高分子物理化学方面的贡 献,几乎遍及各个领域。既 是实验家又是理论家,是高 分子科学理论的主要开拓者 和奠基人之一。著有《高分 子化学原理》和《长链分子 的统计力学》等。

42
第二节 高分子链的近程结构 1立体化学在高分子中的表现 构型——分子中由化学键所固定的原子在空间的几何排列。
要改变构型必须经过化学键的断裂和重组。

43
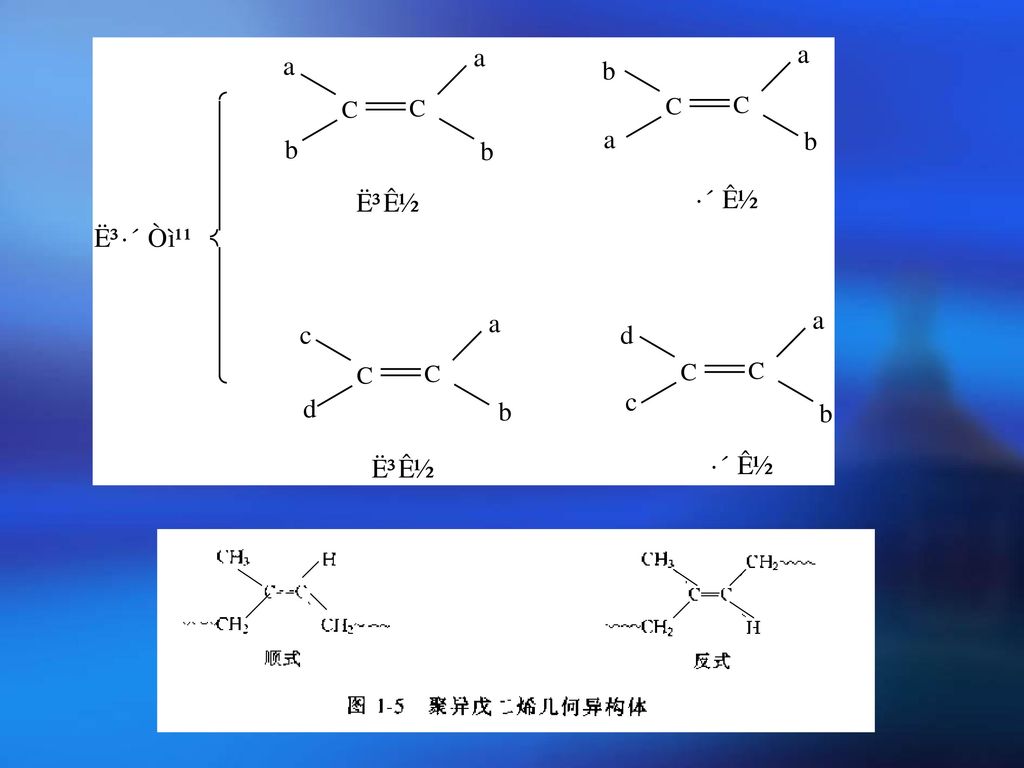
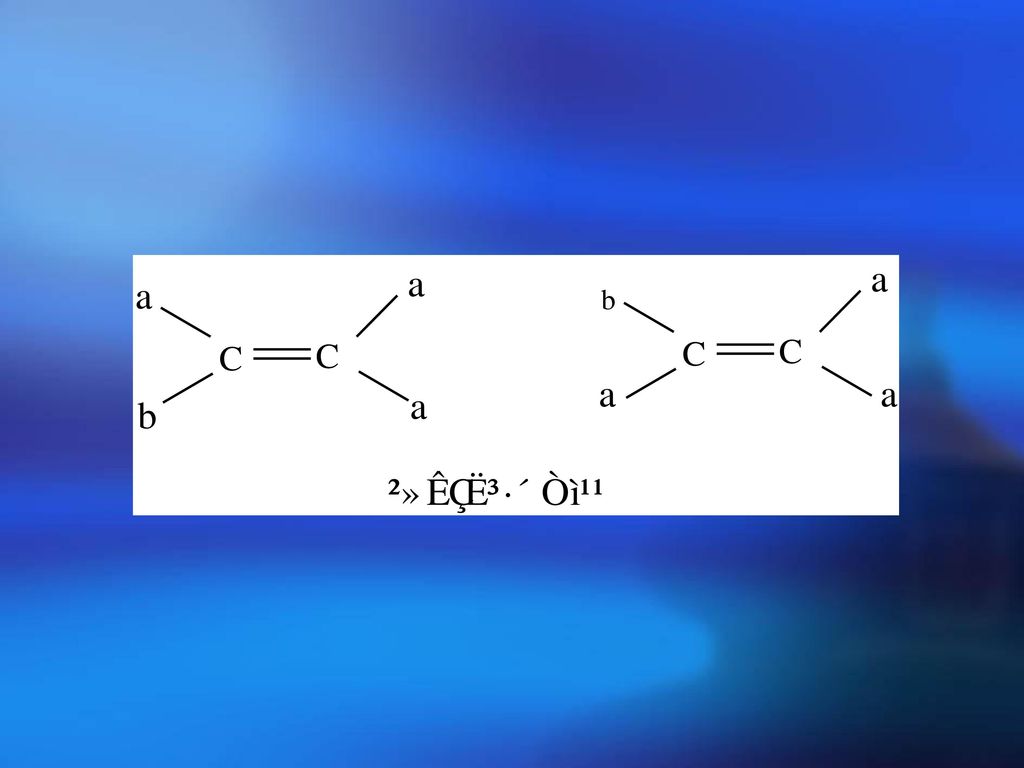
立体异构的分类 几何异构——内双键上的基团在双键两侧排列方式不同而引起的异构(因为内双键中键是不能旋转的)。
。")
46
例如 因为双键上一个C原子上连接二个相同的H,翻个身是同样的化合物。根据定义只有内双键才有顺反异构。

47
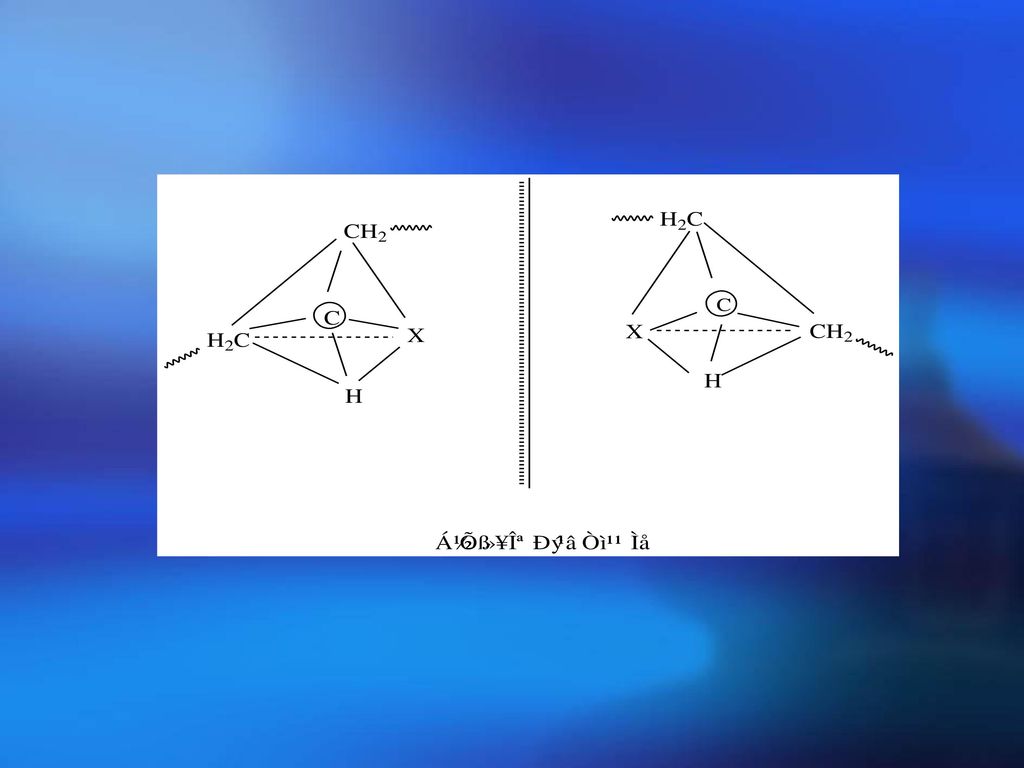
2.立体异构的分类 空间立构——若正四面体的中心原子上四个取代基是不对称的(即四个基团不相同)。此原子称为不对称C原子,这种不对称C原子的存在会引起异构现象,其异构体互为镜影对称,各自表现不同的旋光性,故称为旋光异构。
。此原子称为不对称C原子,这种不对称C原子的存在会引起异构现象,其异构体互为镜影对称,各自表现不同的旋光性,故称为旋光异构。")
48
小分子

49
大分子: 有不对称碳原子,所以有旋光异构

51
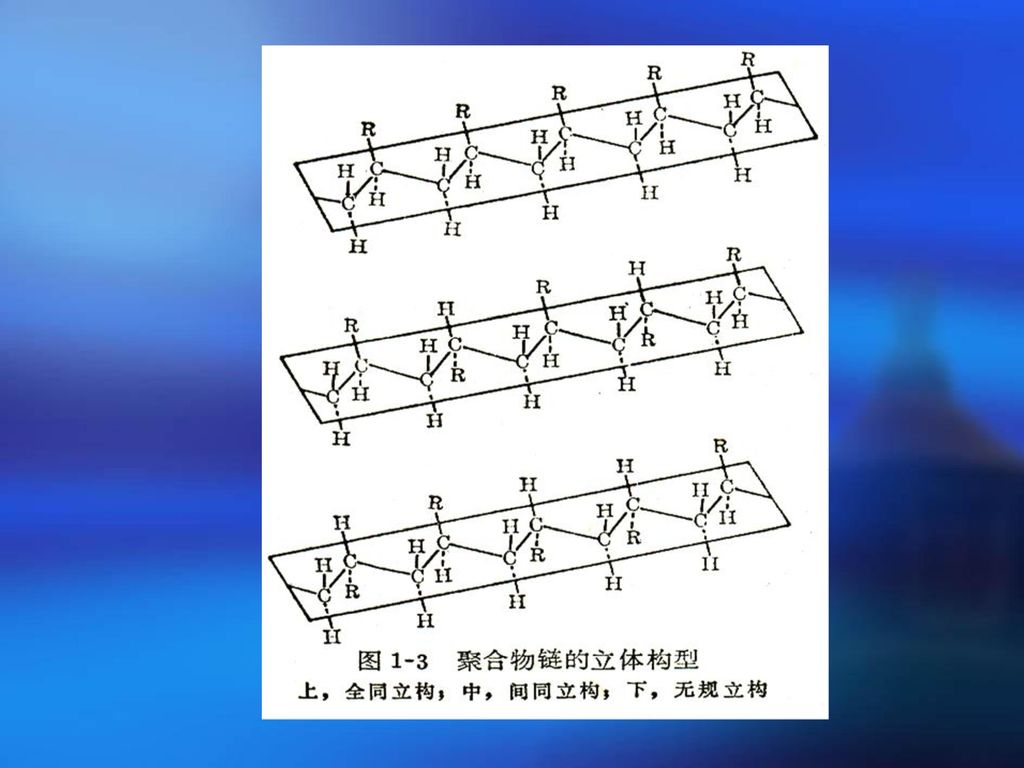
3.三种键接方式 全是由一种旋光异构单元键接而成(全同立构)——取代基全在平面的一侧
由两种旋光异构单元间接键合而成(间同立构)——取代基间接分布在平面两侧 由两种旋光异构单元无规则键合而成(无规立构)——取代基无规则分布在平面两侧
——取代基间接分布在平面两侧. 由两种旋光异构单元无规则键合而成(无规立构)——取代基无规则分布在平面两侧.")
53
举例说明 1. —单烯

54
2.双烯类:丁二烯

55
3. 异戊二烯

56
分子的立体构型不同,导致材料性能差异 PS: 等规PS:规整度高,能结晶, ℃,不易溶解 无规PS:软化点80℃,溶于苯 PP:

57
立体构型表征 等规度(tacticity)——全同立构与间同立构之和所占百分比 立体构型的测定方法(几个纳米) X射线、核磁共振(NMR)、红外光谱(IR)等方法
——全同立构与间同立构之和所占百分比 立体构型的测定方法(几个纳米) X射线、核磁共振(NMR)、红外光谱(IR)等方法")
58
键接异构 头-头结构比例有时相当大。 例如:聚偏氟乙烯,(NMR)10~15% 聚氟乙烯, 6~1 0% 当位阻效应很小及链生长端(自由基、阳离子、阴离子)的共振稳定性很低,得到比较大的头-头或尾-尾
10~15% 聚氟乙烯, 6~1 0% 当位阻效应很小及链生长端(自由基、阳离子、阴离子)的共振稳定性很低,得到比较大的头-头或尾-尾.")
59
4高分子链的支化与交联 大分子链的形式有: 线型(linear) 支化(branching) 网状(network)
 支化(branching) 网状(network)")
60
4-1线型大分子链 一般高分子是线型的。它是由含二官能团的反应物反应的,如前所述的聚氯乙烯和聚酯,分子长链可以卷曲成团,也可以伸展成直线,这取决于分子本身的柔顺性及外部条件。 线型高分子间无化学键结合,所以在受热或受力情况下分子间可以互相移动(流动),因此线型高分子可在适当溶剂中溶解,加热时可熔融,易于加工成型。
,因此线型高分子可在适当溶剂中溶解,加热时可熔融,易于加工成型。")
61
4-2支链形高分子 由于加聚过程中有自由基的链转移发生,常易产生支化高分子。 支化分子对高分子材料的使用性能有一定的影响 下面以PE为例

62
以PE为例 LDPE(Low Density PE)(自由基聚合)
这种聚合方式易发生链转移,则支链多,密度小,较柔软。用于制食品袋、奶瓶等等 HDPE(配位聚合,Zigler催化剂) 这种聚合方法不同与前,获得的是几乎无支链的线型PE,所以密度大,硬,规整性好,结晶度高,强度、刚性、熔点均高。可用作工程塑料部件,绳缆等等 支化度越高,支链结构越复杂则对性能的影响越大,例如无规支化往往降低高聚物薄膜的拉伸度,以无规支化高分子制成的橡胶其抗拉强度及伸长率均比线型分子制成的橡胶为差。
 这种聚合方法不同与前,获得的是几乎无支链的线型PE,所以密度大,硬,规整性好,结晶度高,强度、刚性、熔点均高。可用作工程塑料部件,绳缆等等. 支化度越高,支链结构越复杂则对性能的影响越大,例如无规支化往往降低高聚物薄膜的拉伸度,以无规支化高分子制成的橡胶其抗拉强度及伸长率均比线型分子制成的橡胶为差。")
63
2.支化度的表征 支化度——两相邻支化点之间链的平均分子量来表示支化的程度,称为支化度
支化高分子的形式:星形(Star)、梳形(Comb)、无规(Random)
、梳形(Comb)、无规(Random)")
64
星形 树枝状 短支化 长支化 梯形

65
1-4-3网状(交联)大分子 缩聚反应中有三个或三个以上官能度的单体存在时,高分子链之间通过支链联结成一个三维空间网形大分子时即成交联结构
交联与支化有本质区别 支化(可溶,可熔,有软化点) 交联(不溶,不熔,可膨胀)
 交联(不溶,不熔,可膨胀)")
66
交联高分子的表征 交联度:用相邻两个交联点之间的链的平均分子Mc 来表示。交联度越大,Mc越小。
交联点密度:交联的结构单元占总结构单元的分数,即每一结构单元的交联几率。

67
应用: 橡胶硫化就是在聚异戊二烯的分子间产生硫桥

68
S S S S S S CH2-CH-CH-CH2 -CH2-CH=CH-CH2 -CH2-CH-CH-CH2 S S S S S S S 互接网络示例:硫化橡胶 S S S S S CH2-CH-CH-CH2 -CH2-CH=CH-CH2 -CH2-CH-CH-CH2 S S S S S S S S S S S S CH2-CH-CH-CH2 -CH2-CH=CH-CH2 -CH2-CH-CH-CH2 S S S S

69
应用 另外一种交联PE,它是经过辐射交联,使得软化点和强度均大大提高,大都用于电气接头,电缆的绝缘套管等
除无规交联外,还有规整的网络结构,如:耐高温的全梯型吡隆,耐高温的碳纤维。

70
线型、支化、网状分子的性能差别 线型分子:可溶,可熔,易于加工,可重复应用,一些合成纤维,“热塑性”塑料(PVC,PS等属此类)
支化分子:一般也可溶,但结晶度、密度、强度均比线型差 网状分子:不溶,不熔,耐热,耐溶剂等性能好,但加工只能在形成网状结构之前,一旦交联为网状,便无法再加工,“热固性”塑料(酚醛、脲醛属此类)
")
71
5 共聚物(copolymer) 如果高分子由两种以上的单体组成,则高分子链的结构更加复杂 将有序列分布问题
 如果高分子由两种以上的单体组成,则高分子链的结构更加复杂 将有序列分布问题")
72
两种或多种单体共聚时,结构单元之间连接的序列结构更为复杂。具体可分为无规共聚、交替共聚、接枝共聚、嵌段共聚等。

73
均聚物 无规 嵌段 交替 接枝

74
5-1 无规共聚(random) 两种高分子无规则地联结 ABAABABBAAABABBAAA
由于两种高分子平行无规则地排列改变了结构单元的相互作用,也改变了分子间的相互作用,因此在溶液性质、结晶性质、力学性质方面和均聚物有明显不同。

75
例1: PE,PP是塑料,但 乙烯与丙烯无规共聚的产物为橡胶。
例2: PTFE(聚四氟乙烯)是塑料,不能熔融加工,但四氟乙烯与六氟丙烯共聚物是热塑性的塑料。
是塑料,不能熔融加工,但四氟乙烯与六氟丙烯共聚物是热塑性的塑料。")
76
5-2 嵌段共聚(block) AAAAAABBBBBAAABBBBAAAAA
例如用阴离子聚合法制得的SBS树脂(牛筋底)就是苯乙烯与丁二烯的嵌聚共聚物,其分子链的中段是聚丁二烯(顺式),两端是聚苯乙烯。
就是苯乙烯与丁二烯的嵌聚共聚物,其分子链的中段是聚丁二烯(顺式),两端是聚苯乙烯。")
77
SBS:在120℃可熔融,可用注塑成形,冷到室温时,由于PS的玻璃化转变温度高于室温,分子两端的PS变硬,而分子链中间部分PB的玻璃化转变温度低于室温,仍具有弹性,显示高交联橡胶的特性。SBS不是用化学键交联,而是通过玻璃态PS “交联”的,这是物理交联。

78
顺式聚丁二烯在常温下是一种橡胶,而不是硬性塑料,两者是不相容的,因此SBS具有两相结构:聚丁二烯(PB)易形成连续的橡胶相,PS易形成微区分散区树脂中,它对PB起着交联的作用,PS是热塑性的(thermoplastic),在高温下能流动,SBS是一种可用注塑方法进行加工,而不需要硫化的橡胶,又称为热塑性弹性体(牛筋底),这是橡胶工业上一个重大进步。
易形成连续的橡胶相,PS易形成微区分散区树脂中,它对PB起着交联的作用,PS是热塑性的(thermoplastic),在高温下能流动,SBS是一种可用注塑方法进行加工,而不需要硫化的橡胶,又称为热塑性弹性体(牛筋底),这是橡胶工业上一个重大进步。")
79
5-3 接枝共聚(graft) ABS树脂(acrylonitrile-butadiene-styrene)是丙烯腈、丁二烯和苯乙烯的三元共聚物,共聚方式上是无规与接枝共聚相结合。
 ABS树脂(acrylonitrile-butadiene-styrene)是丙烯腈、丁二烯和苯乙烯的三元共聚物,共聚方式上是无规与接枝共聚相结合。")
80
ABS 可以是以丁苯橡胶为主链,将苯乙烯和丙烯腈接在支链上;也可以以丁睛橡胶为主链,将苯乙烯接在支链上;也可以以苯乙烯—丙烯睛为主链,将丁二烯和丙烯腈接在支链上。
ABS兼有三种组分的特性:丙烯腈有CN基,使聚合物耐化学腐蚀,提高抗张强度和硬度;丁二烯使聚合物呈现橡胶态韧性,提高抗冲性能;苯乙烯的高温流动性好,便于加工成型,而且可以改善制品光洁度。

81
5-4 交替共聚(alternating) ABABABAB 共聚物往往可改善高聚物某种使用性能
PMMA分子中的酯基有极性,使分子与分子间的作用力比PS大,所以流动性差,不易注塑成型。 MMA+S共聚,改善高温流动性,可注塑成型。 S+AN 冲击,耐热,耐化学腐蚀都有提高,可作耐油的机械零件。

82
5-5 共聚物结构的表征 平均组成:化学法(元素分析,官能团沉淀),光谱法(红外,紫外…)。 组成分布(GPC法)。
序列结构(无规共聚物): 例如A和B两种单体相邻二单元有3种链接方式(AA、BB、AB),相邻三单元则有(AAA、BBB、AAB、ABB、ABA、BAB)6种方式链接。
: 例如A和B两种单体相邻二单元有3种链接方式(AA、BB、AB),相邻三单元则有(AAA、BBB、AAB、ABB、ABA、BAB)6种方式链接。")
83
化学组成 单体单元键合 近程结构 单个高分子链的键接(交联与支化) 单体单元主体构型(空间排列) 高分子链结构 高分子的大小(分子量) 远程结构 高分子的形态(构象) 晶态(Crystalline) 非晶态(Non—crystalline) 高分子聚集态结构 取向态(orientatim) 液晶态(Liquid crystals) 织态(texture)
 单体单元主体构型(空间排列) 高分子链结构 高分子的大小(分子量) 远程结构 高分子的形态(构象) 晶态(Crystalline) 非晶态(Non—crystalline) 高分子聚集态结构 取向态(orientatim) 液晶态(Liquid crystals) 织态(texture)")
84
第三节 高分子的远程结构 (long-range structure)
远程结构的内容包括: 1. 高分子的形态(morphology), 或叫构象(conformation) 2. 高分子的大小,即分子量及其分布
, 或叫构象(conformation) 2. 高分子的大小,即分子量及其分布.")
85
2-1 高分子链的柔顺性(flexibility)
一个典型的线形高分子链长度与直径之比 是很大的。例如聚异丁烯大分子 所以 。这就是说,这个大分子长度是直径的5万倍。这样一根细而长的“网丝”,在无外力作用下,不可能是一条直线,而是自然的曲线。这就使得聚异丁烯大分子有着“柔顺性”,也使聚异丁烯材料有着它独特的“高弹性”。从结构上看,是什么根本原因使得高分子有柔顺性呢?我们要从低分子讲起。

86
2-2-1 低分子的内旋转 从有机中知,C—C,C—O,C—N等单键是 键,其电子云的分布是轴形对称的。因此由键相连的两个原子可以相对旋转(内旋转)而不影响其电子云的分布。单键内旋转的结果是使分子内与这两个原子相连的原子或基团在空间的位置发生变化 例如乙烷:如果C—C发生内旋转,则分子内与C相连的H的相对位置就要发生变化(如下图) 这种由于单键内旋转而产生的分子在空间的不同形态称为构象(conformation)
 这种由于单键内旋转而产生的分子在空间的不同形态称为构象(conformation)")
87
H C 迭同式(顺式)构象最不稳定 交叉式(反式)构象最稳定 或
构象最不稳定 交叉式(反式)构象最稳定 或")
88
视线在C-C键方向两个C原子上的C-H键重合时叫顺式,相差60度角时叫反式。
时为顺式,位能最高。 时为反式,乙烷分子位能最低。如下图:

89
顺 式 反 式 位能 (度) 乙烷的内旋转位能图
 乙烷的内旋转位能图")
90
位垒:从一种构象改变为另一种构象时,能量的差值称为内旋转位垒。
内旋转位垒越高,内旋转越困难。由于反式构象能量最低,所以1,2-二氧乙烷在晶体时绝大部分是反式构象。

91
X Z Y ⑴ ⑶ ⑵ C1 C3 C2 C4

92
一.理想情况下 a.碳链上不带有任何其它原子或基团时,C-C单键旋转是没有位阻效应, C-C单键的内旋转完全是自由的,如上图所示。
b.如果我们把C1-C2键固定在Z轴上,则(1)的自转(内旋转)将带动(2)的公转,由于有C-C和C-C之间键角的限制,所以(2)的轨迹是个圆锥面,所以C3可以出现在这个圆锥面的任何位置上。
的自转(内旋转)将带动(2)的公转,由于有C-C和C-C之间键角的限制,所以(2)的轨迹是个圆锥面,所以C3可以出现在这个圆锥面的任何位置上。")
93
一.理想情况下 c.同理(2)的自转,带动(3)的公转,(3)的轨迹也是圆锥面,C4可以出现在圆锥面的任何位置上。
d.事实上,(1)和(2)同时自转,所以(2)和(3)同时在公转,所以,(4)的活动余地就更大了。 e.一个高分子有许多单键,每个单键都能内旋转,所以高分子在空间的形态有无穷多个。
和(2)同时自转,所以(2)和(3)同时在公转,所以,(4)的活动余地就更大了。 e.一个高分子有许多单键,每个单键都能内旋转,所以高分子在空间的形态有无穷多个。")
94
①(1)键自转带动(2)键公转,(2)键的轨 迹是圆锥面,C3可在圆锥面上出现的位置假定 为m个。 ②(2)键自转带动(3)键公转,(3)键的轨 迹同样是圆锥面,C4可在圆锥面上出现的位置 假定也为m个。 ③当只考虑(2)自转、不考虑它公转时,则C4 有m 个位置可出现,如果也考虑(2)的公转, 则C4就有m2个位置可出现。 ④以此类推,对于第i键上的第i+1个原子来讲, 如果考虑所有键的公转和自转,则他它上面的 原子的出现位置是mi-1个。
键自转带动(2)键公转,(2)键的轨 迹是圆锥面,C3可在圆锥面上出现的位置假定 为m个。 ②(2)键自转带动(3)键公转,(3)键的轨 迹同样是圆锥面,C4可在圆锥面上出现的位置 假定也为m个。 ③当只考虑(2)自转、不考虑它公转时,则C4 有m 个位置可出现,如果也考虑(2)的公转, 则C4就有m2个位置可出现。 ④以此类推,对于第i键上的第i+1个原子来讲, 如果考虑所有键的公转和自转,则他它上面的 原子的出现位置是mi-1个。")
95
第(2)键上(C3)出现的位置为m2-1=m 第(3)键上(C4)出现的位置为m3-1= m2 第(4)键上(C5)出现的位置为m4-1=m3 …… 第(i)键上(Ci+1)出现的位置为
键上(C3)出现的位置为m2-1=m 第(3)键上(C4)出现的位置为m3-1= m2 第(4)键上(C5)出现的位置为m4-1=m3 …… 第(i)键上(Ci+1)出现的位置为")
96
二.实际上 内旋转完全自由的C-C单键是不存在的,因为碳键上总要带有其它原子或基团,当这些原子或基团充分接近时,电子云之间将产生斥力使单键的内旋转受到阻力,所以高分子的形态(构象)也不可能是无穷多的,而是相当多的
也不可能是无穷多的,而是相当多的.")
97
三.“链段”的概念 把高分子链想象为一根摆动着的绳子,它是有许多可动的段落连接而成的,由前面所讲的分析可推想,当i足够大时,链中第i+1个键上的原子在空间可取的位置已与第一个键完全无关了。所以长链可以看作是由许多链段组成,每个链段包括i个键。链段之间可看成是自由连接的,它们有相对的运动独立性,不受键角限制。

98
链段的定义 高分子链上划分出的可以任意取向的最小单元或高分子链上能够独立运动的最小单元称为链段。
所以高分子链上单键数目越多,内旋转越自由,则高分子链的形态(构象)越多,链段数也越多,链段长度越小,链的柔顺性越好。
越多,链段数也越多,链段长度越小,链的柔顺性越好。")
99
高分子的柔顺性的实质 高分子的柔顺性的实质就是大量C-C单键的内旋转造成的。 极端情况:
当高分子链上每个键都能完全自由旋转(自由联接链),“链段”长度就是键长——理想的柔性链(不存在)。 当高分子链上所有键都不能内旋转——理想的刚性分子(不存在),“链段”长度为链长。
, 链段 长度就是键长——理想的柔性链(不存在)。 当高分子链上所有键都不能内旋转——理想的刚性分子(不存在), 链段 长度为链长。")
100
2-2高分子链的柔顺性的定量描述和构象统计理论
2-2-1柔顺性的定量描述: 由于高分子链的内旋转情况复杂,不能像小分子一样用位能的数据来表示柔顺性

101
1.大分子链尺寸的表示方法 表征方法很多,常用的有三种: “链段”长 主要介绍前两种 均方旋转半径

102
1)均方末端距(线型分子) (mean square end to end distance )
当分子是实心球时,可用球半径表示其尺寸。 当分子是细杆,可用杆长和截面半径来表征其尺寸。 当分子是瞬息万变的无规线团状的高分子时,我们可用方均根末端距来表示分子尺寸,如下图:

103
理解: 末端距——线型高分子链的一端到另一端达到的直线距离。这是一个向量,高分子链愈柔顺、卷曲愈厉害,愈小。
均方末端距——由于构象随时在改变,所以末端距也在变化,只好求平均值,但由于方向任意,所以平均值必为零,没有意义。所以先将平方再平均,就有意义了,这是一个标量。

104
2)均方旋转半径 (支化分子) 对于支化聚合物大分子来讲,一个分子有若干个端基。这样均方末端距的意义就不明确了,所以引入新的表征方式
(旋转半径)——从大分子链的质量中心到各个链段的质量中心的距离,是向量 (均方旋转半径)——旋转半径的平方值的平均。是标量,越小越柔顺
——从大分子链的质量中心到各个链段的质量中心的距离,是向量. (均方旋转半径)——旋转半径的平方值的平均。是标量,越小越柔顺.")
Similar presentations











>")





是由30个碳原子组成的萜类化合物。(指基本骨架,不包括糖),可认为是由6个异戊二烯缩合而成的。 分类 从结构上分两大类:四环三萜 五环三萜 存在形式:游离形式(苷元) 苷的形式(与糖结合)>")

