临床药代动力学研究及相关问题 北京协和医院临床药理中心 胡蓓
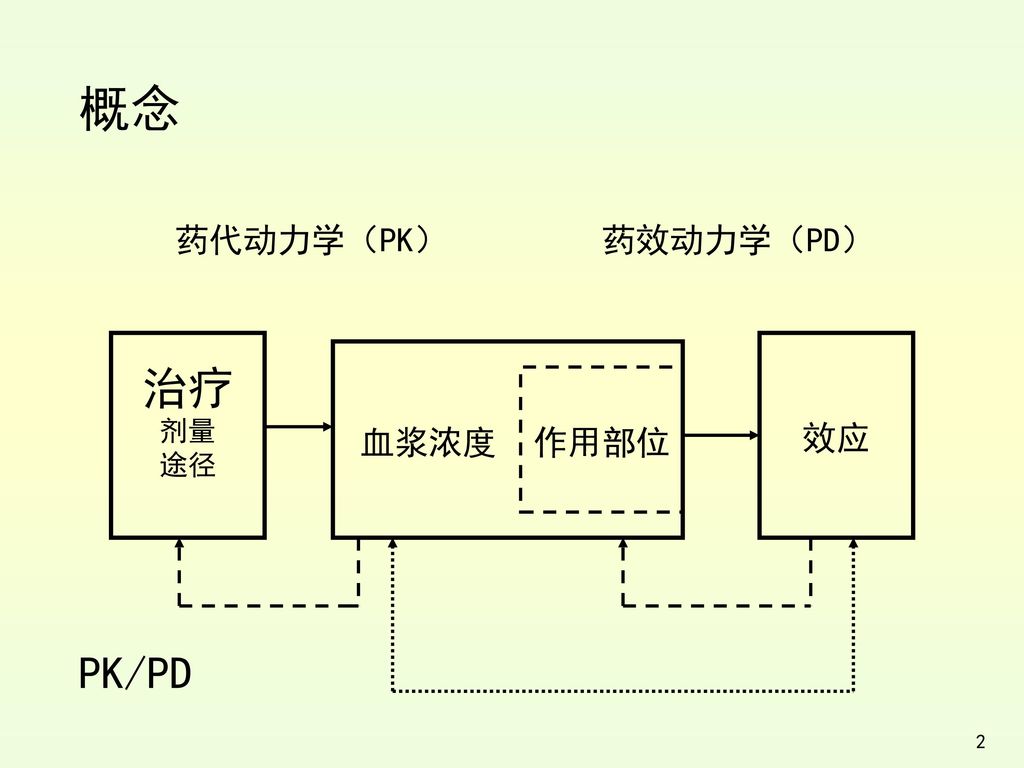
概念 药代动力学(PK) 药效动力学(PD) PK/PD 治疗 剂量 途径 效应 血浆浓度 作用部位
PK基础 定义 药物代谢动力学 = 药物在体内发生了什么
PK基础 定义 吸 收 分 布 消 除 Resorption= Absorption + first pass effect
PK基础 吸收 吸收 = 药物从给药部位转移 到附近的组织
PK基础 吸收 口服进入 例如:药物S 剂量 已不是吸收的问题了 肠壁 肠腔 体循体 吸收 测量部位 代谢 代谢 粪便 小肠 肝 ∴ 生物利用度 = 10% 代谢 代谢 粪便 小肠 肝 首过效应(两部分)
PK基础 吸 收 分 布 消 除
蛋白结合 碱性药物 f (u) = 酸性药物 PK基础 分布 1 a-酸性糖蛋白 白蛋白 n [P] 1+ [K]+[D] [P]:蛋白浓度 1.0 0.8 0.6 0.4 0.2 1 a-酸性糖蛋白 f (u) = 白蛋白 n [P] 1+ 酸性药物 [K]+[D] [P]:蛋白浓度 n : 固定作用部位的数量 [K]:亲和常数 [D]:药物浓度 10-6 0.25 10-5 2.5 10-4 25 10-3 250 10-2 2500 药物浓度 相比于白蛋白, a-酸性糖蛋白更容易被饱和
PK基础 吸 收 分 布 消 除
PK基础 消除 代谢 吸收和分布 Ⅰ相代谢 例:CYP酶-微粒体 Ⅱ相代谢 例:磺基转移酶-胞浆 排泄
PK基础 消除 代谢 化合物 酶 毒 性 活动 Ⅰ相代谢产物 酶 Ⅱ相 代谢产物
代谢 PK基础 消除 肝代谢,还有肠、肺、血液… 代谢 = 酶类 抑制、诱导、互动 基因、年龄、环境的影响 区域内和区域间的个别差异性 冬夏食物不同诱导可能不同 CYP3A 43% CYP2A6 2% CYP2C19 4% CYP2D6 30% CYP2E1 5% CYP1A2 6% CYP2C9 10% P450所涉及的药物代谢%
PK基础 消除 肠肝循环 肝 胆汁 肠 药物和代谢物的消除
尿排泄的机制 4. 肾小球过滤 + 分泌物 + 重吸收 PK基础 消除 0<CLR<625ml/min -在pH = 6环境下非离子化的亲脂性化合物
PK基础 参数 不同剂型采用不同的给药途径 血液样品 尿,粪便,胆汁…
PK基础 参数 Cmax 浓度 吸收(主要的) 分布 消除 时间 Resorption + +
PK基础 参数 分布 + 消除 浓度 纯粹消除 Ci Ci/2 t 1/2 时间
PK基础 参数 浓度 = 药物暴露的评估 AUC =曲线下面积 时间
I期临床药代动力学研究的方案设计 SFDA指导原则 根据药物的特点设计研究
制定I期临床研究方案经常遇到的问题 如何确定起始剂量 如何进行剂量分组 如何确定最大耐受剂量 为什么要进行单剂给药的药代动力学研究 为什么要进行多剂给药的药代动力学研究 是否需要进行特殊人群的药代动力学研究 是否需要进行代谢产物的药代动力学研究
制定I期临床研究方案经常遇到的问题 起始剂量:来自动物实验未观察到不良反应的 剂量〔no-observed-adverse-effect-level (NOAEL)〕换算成人体剂量后的一个分量 (e.g.,half log or less)。
I期研究起始剂量的确定 确定起始剂量的原则:安全、科学 确定起始剂量参考的文献:Guidance for Industry and Reviewers Estimating the Safe Starting Dose in Clinical Trials for Therapeutics in Adult Healthy Volunteers (FDA, CDER) FDA 对于I期临床研究提出了人体等效剂量HED (human equivalent dose)的概念,从动物实验数据推算可能产生等价药效的人体剂量。
Pharmacology and Toxicology Guidance for Industry Estimating the Maximum Safe Starting Dose in Initial Clinical Trials for Therapeutics in Adult Healthy Volunteers U.S. Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation and Research (CDER) July 2005 Pharmacology and Toxicology
Db=Da.Rab(Da和Db是标准体重剂量,mg.kg-1 , Rab是换算系数) 举例:I期研究的起始剂量确定 Db=Da.Rab(Da和Db是标准体重剂量,mg.kg-1 , Rab是换算系数) 动物品种 小鼠b 大鼠b 比格犬b 成人b 标准体重/kg 0.02 0.15 10.0 60.0 表面积/m2 0.0066 0.025 0.5 1.62 体重系数 0.0898 0.0886 0.1077 0.1057 系数S 3 6 20 37 小鼠a 1.00 0.500 0.150 0.081 大鼠a 2.00 0.300 0.162 比格犬a 6.67 3.33 0.541 成人a 12.33 6.17 1.85 例:已知150g(标准体重)大鼠用5mg.kg-1,求成人(标准体重)的用药剂量:查表,大鼠a行,成人b列的Rab=0.162,故成人的剂量为: Db= Da.Rab=5*0.162=0.81mg.kg-1。 引自:www.cncro.com
抗肿瘤药物临床试验技术指导原则(第二稿) 二〇〇七年三月 尚未颁布 举例:抗肿瘤药物的起始剂量确定 多数抗肿瘤药物的治疗指数很窄,较高的起始剂量可能导致出现严重毒性,甚至患者死亡,从而使得原本具有很好潜力的有效药物不能得以继续研发。另一方面,如果选择过低的起始剂量,那么就有可能使得试验周期延长,造成资源浪费,而且从伦理学角度考虑,不应使过多患者暴露在无效剂量下。因此,起始剂量的选择应当综合非临床药效、毒理和药代动力学/毒代动力学的研究结果综合考虑。 抗肿瘤药物临床试验技术指导原则(第二稿) 二〇〇七年三月 尚未颁布
抗肿瘤药物临床试验技术指导原则(第二稿) 二〇〇七年三月 尚未颁布 举例:抗肿瘤药物的起始剂量确定 对于细胞毒类药物,I期临床试验的起始剂量计算原则上相当于非临床试验中啮齿类动物MTD剂量的1/10,或非啮齿类动物MTD剂量的1/6,单位用mg/m2表示,同时还需考察MTD剂量在其他种属动物的毒性反应及可逆性。具体可参考《细胞毒类抗肿瘤药非临床研究指导原则》。 抗肿瘤药物临床试验技术指导原则(第二稿) 二〇〇七年三月 尚未颁布
抗肿瘤药物临床试验技术指导原则(第二稿) 二〇〇七年三月 尚未颁布 举例:抗肿瘤药物的起始剂量确定 对于一些非细胞毒类抗肿瘤药,由于其毒性相对较小,I期临床试验的起始剂量计算可采用非临床试验中非啮齿类动物NOAEL(未观察到不良反应的剂量)的1/5,或者更高。 若为国外已进行临床试验的新化合物,已有可靠的可借鉴临床试验资料,参照国外临床研究数据设计国内临床试验的起始剂量也是可以接受的。此时应当考虑不同人种间的差异可能带来的影响。 抗肿瘤药物临床试验技术指导原则(第二稿) 二〇〇七年三月 尚未颁布
抗肿瘤药物临床试验技术指导原则(第二稿) 二〇〇七年三月 尚未颁布 举例:抗肿瘤药的剂量递增 耐受性试验:剂量间隔可先大后小 举例:改良Fibonacci 固定百分数递增法,即确定初试剂量后,第二剂量应比第一剂量多一倍;第三剂量比第二剂量多67%;第四剂量比第三剂量多50%;第五剂量比第四剂量多40%;以后的各级均应比上一级多33%。 对于细胞毒药物,剂量逐渐递增到MTD就可停止爬坡。 抗肿瘤药物临床试验技术指导原则(第二稿) 二〇〇七年三月 尚未颁布
抗肿瘤药物临床试验技术指导原则(第二稿) 二〇〇七年三月 尚未颁布 举例:抗肿瘤药的剂量递增 有些非细胞毒类药物的毒性很小,可能不能观察到明显的MTD。但即使药物活性的靶点已经饱和或在没有显著毒性的时候就观察到了明显疗效,也仍然建议研究更高的剂量,以便更好的明确化合物的安全性。如果剂量递增到观察到疗效后,继续增加剂量并没有看到疗效的增加,而毒性增加明显,则应选择较低的剂量进行下一步的研究。 抗肿瘤药物临床试验技术指导原则(第二稿) 二〇〇七年三月 尚未颁布
I期研究方案的设计问题 常见的问题是:剂量间隔过大导致安全性问题或I期试验未达到最大耐受剂量,限制了II期临床试验的剂量范围,未能达到有效剂量。 如何确定最大耐受剂量? 遇到不良事件是否立刻停止试验? 已达到方案预先设计的最大耐受剂量是否停止试验? 毒性分级表(轻、中、重、危及生命) 推荐使用毒性分级表
Toxicity Grading Scale for Healthy Adult and Adolescent Volunteers Guidance for Industry Toxicity Grading Scale for Healthy Adult and Adolescent Volunteers Enrolled in Preventive Vaccine Clinical Trials U.S. Department of Health and Human Services Food and Drug Administration Center for Biologics Evaluation and Research April 2005
Potentially Life Threatening (Grade 4) 举例:FDA毒性分级表 Mild (Grade 1) Moderate (Grade 2) Severe (Grade 3) Potentially Life Threatening (Grade 4) ALP 1.1-2.0 x ULN 2.1-3.0 x ULN 3.0-10 x ULN > 10 x ULN ALT, AST 1.1-2.5 x ULN 2.6-5.0 x ULN 5.1-10 x ULN Bilirubin* 1.1-1.25 x ULN 1.26-1.5 x ULN 1.51-1.75 x ULN > 1.75 x ULN *when accompanied by any increase in Liver Function Test
抗肿瘤药物临床试验技术指导原则(第二稿) 二〇〇七年三月 尚未颁布 举例:抗肿瘤药的毒性反应观察和评价 不良反应性质和严重程度的评价标准遵照国际上通用的药物毒性反应标准(美国国立癌症研究所[NCI]的常见毒性反应标准(Common Toxicity Criteria ,CTC)进行。 抗肿瘤药物临床试验技术指导原则(第二稿) 二〇〇七年三月 尚未颁布
抗肿瘤药物临床试验技术指导原则(第二稿) 二〇〇七年三月 尚未颁布 举例:抗肿瘤药的剂量递增 为避免更多受试者使用无效药物,在每一剂量水平应选用尽量少的可达到评价要求的患者,一般至少有3名或3名以上可评价的受试者,但若某一剂量并无毒性或很小毒性反应,少于3名受试者也是可接受的。若出现明显毒性,应考虑增加受试者例数。如某一剂量组有1例产生3度以上不良反应,则该剂量水平应继续增加3例受试者,如不再出现,可进入下一剂量组,如仍出现,则停止剂量爬坡。只有当特定剂量水平获得足够评价资料后方可进入下一个剂量水平。 抗肿瘤药物临床试验技术指导原则(第二稿) 二〇〇七年三月 尚未颁布
抗高血压药物临床试验技术指导原则(第二稿) 二〇〇七年三月 尚未颁布 举例:抗高血压药的耐受性研究 应该进行单次和多次给药的人体耐受性研究,研究中可以同时观察试验药物的降压效应、主要不良反应的类型和程度等,试验中需要制定明确的终止标准。 抗高血压药物临床试验技术指导原则(第二稿) 二〇〇七年三月 尚未颁布
I期临床试验中耐受性试验与 药代动力学的关系 单独的耐受性试验 与耐受性试验结合的药代试验 Intensive 药代试验
SFDA指导原则:临床药代动力学试验 健康受试者的药代动力学 单次给药(线性药代动力学研究) 多次给药(稳态药代动力学研究) 食物的影响 药物 – 药物相互作用 代谢产物的PK
SFDA指导原则:临床药代动力学试验 特殊人群的药代动力学 肝功不全 肾功不全 老年人 儿童 其他:患者及不同种族
抗高血压药物临床试验技术指导原则(第二稿) 二〇〇七年三月 尚未颁布 举例:抗高血压药的药代动力学研究 “应该进行详细的人体药代研究,抗高血压药物常用于老年人,因此要对老年人进行特殊研究,对于不同程度的肝肾功能不全患者也应进行特殊研究。高血压药物常与其他药物合并使用,应该进行相关药物的药代相互作用研究。同时进行药代/药效研究,可以提供额外的信息。” 抗高血压药物临床试验技术指导原则(第二稿) 二〇〇七年三月 尚未颁布
药代动力学临床试验的设计 单剂给药(线性)药代动力学研究 开放、随机、交叉、单剂、多周期试验设计 开放、随机、交叉、单剂、多周期试验设计 双盲、随机、交叉、单剂、多周期试验设计 开放、随机、平行、单剂、单周期试验设计 双盲、随机、平行、单剂、单周期试验设计
药代动力学临床试验的设计 多剂给药(稳态)药代动力学研究 开放、随机、平行、多剂给药试验设计 双盲、随机、平行、多剂给药试验设计
药代动力学临床试验的设计 食物对药代动力学的影响 开放、随机、交叉、单剂、两周期试验设计 开放、随机、平行、单剂、两阶段试验设计
药代动力学临床试验的设计 特殊人群的药代动力学研究:设计试验时有特殊考虑 肝功能不全患者的药代动力学 肾功能不全患者的药代动力学 肝功能不全患者的药代动力学 肾功能不全患者的药代动力学 老年受试者的药代动力学 儿童受试者的药代动力学
药代动力学临床试验的设计 受试者的数目 SFDA指导原则:至少8例 一般不需要根据统计学的把握度进行计算
药代动力学临床试验的设计 受试者的性别 SFDA指导原则:男女各半(在使用盲法时可能出现数目不等的情况)
药代动力学临床试验的设计 受试者的入选剔除标准 根据SFDA指导原则 因试验方案而异
药代动力学临床试验的设计 取血点的设计(生物等效性试验的要求与此一致) SFDA指导原则:至少9个点 避免第1个取血点是Cmax 在消除相应至少有3个取血点 取血至3-5个消除相半衰期或血药浓度降至 Cmax的1/10-1/20。
药时曲线与采样时间的关系 Conc. tmax Time 标准曲线上限 Cmax 标准曲线下限浓度太高 Cmin<1/10~1/20 结束采样时间太早 AUC0-t 开始采样时间合适 ≥ 80% 开始采样时间太晚 AUC0-∞
I期研究方案的设计问题 单剂药代动力学研究的目的 线性药代动力学的判断方法 为什么要设置至少3个剂量组 相关分析法 等效性分析法 Power模型
药代动力学临床试验实例1 SNF的单次给药药代动力学研究 Single dose: 25, 50, 100mg three way cross-over study
Study Design Day 1 Day 8 Day 15 25mg 50mg 100mg 50mg 100mg 25mg 100mg Group 1 25mg 50mg 100mg Group 2 50mg 100mg 25mg Group 3 100mg 25mg 50mg On each dosing day, 7ml of blood from the vein of forearm were to be taken pre-dose and 0.25, 0.5, 1, 1.5, 2, 3, 4, 6, 8, 10, 12, 16, 24 hours post-dose.
Concentration-time curve of SNF 100 200 300 400 500 600 700 800 900 4 8 12 16 20 24 Time (hr) ng/ml 25 mg 50 mg 100 mg
Latin Square 设计 存在:ABC, BCA,CAB 缺少:CBA, ACB, BAC 试验设计不是很均衡。
双拉丁方设计 仍然12位受试者。分为6组,每组2人。 试验设计更加均衡。有利于减少给药顺序和试验周期的影响。
Williams 设计 组号 1 2 3 4 A B C D
药代动力学参数的估算 将试验中测得的各受试动物的血药浓度-时间的数据分别进行药代动力学参数的估算,求得新药的主要药代动力学参数,其中口服给药包括:Ka(吸收速率常数)、Tmax(峰时间)、Cmax(峰浓度)、AUC(血药浓度-时间曲线下面积)、Vd(表观分布容积)、Kel(消除速率常数)、t1/2(消除半衰期)、CL(清除率)等。静脉注射包括:t1/2(a)、t1/2(b)、K12、K21、K10、Vd、CL(T)、AUC等。
药代动力学参数的估算 可用模型法求算,也可按非房室模型分析。如用电子计算机程序处理数据应指明所用程序的名称和版本。
线性范围的评价方法 1. 以每个动物的参数为基础,通过给药剂量与各个动物的AUC和Cmax的相关分析,得到直线回归方程的相关系数,再对相关系数进行t检验或r检验。检验结果为统计学显著相关时,认为在被检验的剂量范围内药物的机体暴露与剂量的增加成比例。
线性范围的评价方法
线性范围的评价方法
线性范围的评价方法 2. 采用生物等效分析的方法将不同剂量组的、归一化的、经对数转换的Cmax 和 AUC与其中一个剂量组的相应参数进行比较,计算其比值(F)的90%置信区间,以该区间在0.7-1.43 (Cmax)和0.8-1.25 (AUC)之间为等效,并且判断Cmax 和 AUC的增加与剂量的增加成比例。如F值的90%置信区间在0.7-1.43(Cmax)和0.8-1.25(AUC0-∞)之外则判断Cmax 和 AUC的增加与剂量的增加不成比例。
线性范围的评价方法 AUC0-∞ Cmax Mean* 90% CI+ 1.10 1.02-1.19 1.08 0.92-1.25 1.13 10mg:5mg 1.10 1.02-1.19 1.08 0.92-1.25 15mg:5mg 1.13 1.04-1.21 0.93 0.76-1.09 CVw(%)# 10.24 19.42 *= geometric mean += Lower limit-Upper limit #= within-subject coefficients of variation = SQRT(exp(MSE)-1) x 100, where MSE is the residual error from the ANOVA
线性范围的评价方法 3. 采用Power模型(Gough等 1995年提出)分析PK参数(如Cmax 和AUC0-t)与剂量之间的相关关系。其定义为: Ln(PK) = Ln() + * Ln(Dose) + error. 这里假设PK参数和剂量水平的关系为: PK = * Dose^()
线性范围的评价方法 参数被称为比例常数(proportionality constant),它和剂量水平与PK参数水平之比相关。参数被称为模型的形状参数(shape parameter),因为它定义了剂量与PK参数的相关关系曲线的形状。应注意的是,PK参数的剂量比例只依赖于参数值。如果参数值接近于1,则剂量比例能够确立。相反,如果参数值接近于0,则PK参数与剂量相互独立,不存在比例关系。
线性范围的评价方法 PK = * Dose^() =0 =1 =2 Dose PK
线性范围的评价方法 在实际应用中,将不同剂量组的、归一化的、经对数转换的Cmax 和 AUC进行方差分析。使用SPSS软件时,将经对数转换的药代参数作为因变量,受试者为随机因子、周期为固定因子,经对数转换的剂量(ln Dose)为协方差变量,选择GLM单因素方差分析(General Linear Model_Univarite)。显著性水平设为0.1。运算之后程序可以给出ln Dose 的系数,即形状参数。程序亦可以给出其90%的置信区间,如在0.8-1.25的范围至内,可认为剂量比例能够确立。
线性范围的评价方法
多剂药代动力学研究 Cssmax Cssmin Cav Time Conc.
给药间隔等于T1/2 首次剂量是维持剂量的1-3倍
SNF的多次给药药代动力学研究 100mg, multiple-dose study
Study Design n=11 DAY 1 2 3 4 5 6 7 Dose 100mg PK Trough & peak concentration PK On day 1 and day 7, 7ml of blood from the vein of forearm were to be taken pre-dose and 0.25, 0.5, 1, 1.5, 2, 3, 4, 6, 8, 10, 12, 16, 24 hours post-dose.
Concentration-time curve of SNF 200 400 600 800 1000 4 8 12 16 20 24 Time (hr) ng/mL Day 1 Day 7
Drug X 多次给药药代动力学研究 Study Design: Subjects received drug X 1mg every 6 hours at 08:00, 14:00, 20:00 and 02:00 for 48 hours (Days 2 and 3). 29 blood PK samples for each subject were taken within 24 hours after 8am dosing on day 3.
Concentration-time curve of X
SFDA指导原则:临床药代动力学试验 药代/药效动力学链式研究 进行PK/PD Link研究的先决条件 根据新药本身考虑是否有必要 根据新药本身考虑是否有必要 是否有合适的PD评价指标 特殊的软件
药代/药效动力学链式研究 药物浓度时曲线 药效时间曲线 给药x 给药x PK/PD 建模 链式模型 浓度-效应 ‘机体对药物的作用 药物对机体的作用 给药x 给药x PK/PD 建模 链式模型 浓度-效应
药代/药效动力学链式研究 链式模型 - 链式模型的定义 链接PK模型到PD模型 - 很多不同的链式模型,但可归为: 链接浓度到效应 浓度和效应的直接链接,直接链式模型 间接链式模型,时间延迟
药代/药效动力学链式研究 直接链式模型 定义 药时曲线伴随药效时间曲线 在Cmax处观察到Emax (最大效应) 药物浓度下降的同时效应强度下降
药代/药效动力学链式研究 Emax 模型 Emax=efficacy, max response 假设在血浆浓度和效应可直接链接 直接意味着浓度 CA 立即给出效应 EA : 无时间延迟 (相似的tmax), 因此血浆浓度Cp反映了作用部位的浓度 定义 Emax 为最大效应 (依赖于系统的效应) EC50 : 给出50%Emax 时的血浆药物浓度 Emax=efficacy, max response Emax/2 EC50 = potency
药代/药效动力学链式研究 由Emax模型引申出的效应模型 - 最常见的引申模型 Emax达到100 %的Emax模型 线性模型 Sigmoidal模型或 sigmoidal Emax或 HILL模型 0.0 0.2 0.4 0.6 0.8 1.0 1 2 3 4 5 CONCENTRATION (MULTIPLES OF C50) EFFECT (FRACTIONS OF Emax) g = 0.5 g = 5 g = 2 g = 1 理论上, g 是一个整数 如果C = C50,不论 g为何,E = Emax/2 当 g = 1时,双曲线是特殊的sigmoidal 模型 如果 g > 1, 关系是先凹(C < C50),后凸(C > C50),总体上是S形曲线关系 (在线性坐标时)。
药代/药效动力学链式研究 间接链式模型 定义 药时曲线不伴随药效时间曲线 在Cmax之后观察到Emax (最大效应) 药物浓度下降快于效应强度下降
药代/药效动力学链式研究 间接链式模型 定义 药时曲线不伴随药效时间曲线 在Cmax之后观察到Emax (最大效应) 药物浓度下降快于效应强度下降 滞后环 典型的间接模型
间接链式模型 效应隔室方法 Made popular by SHEINER (1979). 1. 在血浆浓度Cp和效应部位之间引入一级吸收动力学, k1e和ke0是微小的常数 2. 估计参数使Ce与效应同步 3. 给出Ce的直接浓度-效应关系 k1e ke0 PK model for Cp Effect site Ce
间接链式模型 效应隔室方法 - 假设在血浆浓度和效应之间间接链接 - 间接链接意味着CA给出效应EA : 定义 Emax :为最大效应 存在一个时间的延迟 因此血浆浓度 Plasma Cp不能反映作用部位的浓度 定义 Emax :为最大效应 EC50 :给出50%Emax 时的受体部位药物浓度 Ke0 : 在血浆和受体之间的平衡常数
间接链式模型 效应隔室方法 定义 效应部位的浓度(Ce)的确伴随效应时间曲线变化 在Cmax出现时观察到Emax (最大效应) Ke0 : 在 Cp 与效应之间校正时间延迟 Ce Cp 在Ce 与效应之间存在直接关系
举例1: S1 研究药物 S1: 在口服和静脉给药后建立S1的PK/PD 模型
举例1: S1 研究目的 研究静脉和口服给药后S1的 PK/PD 特征。 49/102
举例1: S1 PD 参数或生物标志物的选择 Biomarker X Biomarker X 随给药浓度的升高而降低
举例1: S1 研究方法 药代动力学 PK/PD 建模 PK 采样时间 = PD 测量时间: 药效动力学 : 已知作用机制为直接关系(临床终点) IV 和 PO 数据共同建模 比较两种给药途径的PD参数
PK/PD 建模 举例1: S1 PK/PD 模型 : 与受体的直接关系 抑制性 sigmoidal Emax 模型: 随血浆浓度(Cp)变化与基线值相比的降低 E0 : Biomarker X 基线值,Imax : 给药后与基线相比最大抑制百分比,IC50 : 达到最大抑制效应时的浓度, : Sigmoidicity 因子,Cp : 血浆药物浓度
举例1: S1 研究方法 在静脉和口服给药后,在 Biomarker X 和 S1 血浆浓度之间PK/PD关系
举例1: S1 结果: IV / PO的比较 50 100 150 200 250 1 10 1000 10000 100000 S1 (ng/ml) VEB (Beats/min) Observed data after PO adm PKPD model after PO adm Observed data after IV adm PKPD model after IV adm
举例1: S1 结论 - S1 的浓度与Biomarker X 直接相关 - 最大效应约为 90% 几乎恢复正常: 疗效好 - 在静脉给药和口服给药之间,EC50 不同 存在一个或几个活性代谢产物的征兆,且浓度在口服给药之后 >> 静脉给药之后 典型地由口服给药后的首过效应代谢所致Biomarker X
与II期给药方案关系密切的 药代参数 Cmax Tmax 半衰期 线性药代剂量范围 蓄积比 PK/PD & Pop PK
I期研究数据如何支持后续研究 ICH E3 结果的分析是许多研究报告中缺乏的,研究者应该对研究结果逐一进行分析。例如: 可耐受剂量范围与有效浓度的关系 Cmax、Cav与有效浓度的关系 Tmax与起效时间的关系 T1/2、AUC与给药频率的关系 性别、特殊人群、药物-药物相互作用等因素与剂量 调整的关系 ICH E3
I期研究质量对后续研究的影响 安全性数据的收集与评价:是否继续开发 PK数据的准确性:给药方案是否正确 产品说明书的内容
I期研究的目的与作用 I期临床研究的目的:初步的临床药理学及人体安全性评价试验。是在人体上进行新药研究的起始期,为制定给药方案提供依据。 I期临床研究可为后续研究提供以下数据(可覆盖将来药品说明书的大部分内容): 人体最大耐受剂量(MTD)---耐受性试验 线性药代动力学范围(Cmax、AUC、Tmax、t1/2)---单剂PK研 究 稳态参数(蓄积比与波动系数)---多剂PK研究 有效剂量范围---PK/PD Link 服法---进食对口服吸收的影响、半衰期对给药次数的影响 剂量调整---特殊人群用药指导、药物-药物相互作用、代谢产物PK 不良事件 ★说明书★
I期临床药代动力学研究方案实施 I期临床研究室 组织结构 硬件设施 质量控制 人员培训
I期临床试验研究室的 组织结构与管理 以北京协和医院I期临床研究室为例
SOP和质量保障 第一章: I期临床研究室的SOP 第二章: 质量保障体系的SOP 第三章: 生物分析实验室的SOP 各种工作表单:196个 Updated: Oct 24, 2008
I期试验三大关注重点 药品管理/样品管理/急救技术支撑 临床试验药品及各类标准品均有专人专柜管理,严格执行SOP 临床试验生物标本严格按照方案和SOP采集、转运、保管 凡涉及试验药品/生物标本摆放、储存的地方均有24小时全天候环境参数(温度/湿度)测定,人工记录和计算机自动记录对此提供双重保障 与急诊科长期合作,急诊科医师定期为I期病房医护人员培训急救技术,所有I期试验急诊大夫均在试验现场随时提供急救技术支撑
I期临床研究室使用的软件 样本量估计软件 (NQUERY ADVISOR 7.0) 药代分析软件(WinNonlin Enterprise version) 统计软件(SAS) I期病房信息管理:Promasys 实验室信息管理:Watson LIMS
国内、国际认证 在临床药理专业中我室的生物分析实验室于2005年在国内率先获得国际质量体系ISO/IEC 17025认证 获得国家认证认可委员会的计量认证
人员培训情况 自2005年以来每人每年均有年度培训计划 科室负责提供各种内部和外部的GCP/SOP等质量培训及试验技术培训,由质量保障组监督执行 仅2007年一年我室就先后组织了18次质量培训,38次技术培训,总计56次培训。 每次培训均有包括培训时间、地点、内容、培训者、参加者等信息的书面记录
I期临床试验病房的仪器设备 包括48台床边监护仪在内的各类临床设 备168台套 EDC系统1套 扩音系统1套 环境温度湿度监控系统1套
生物分析实验室的仪器设备 包括4台LC-MS/MS在内的各类分析设备39 台套 实验室信息管理系统1套(Watson LIMS) 实验室温度湿度监控系统1套
总结报告撰写 SFDA 指导原则 ICH 指导原则
化学药物临床试验报告的结构与内容技术指导原则 二○○五年三月 已颁布 SFDA 指导原则:1.5.4 讨论和结论 对临床研究的有效性和安全性结果进行总结,讨论并权衡受试药的利益和风险。不要简单地重复结果,也不要引出新的结果。结论应清晰明确,对其意义和可能的问题应结合文献加以评述,阐明对个体患者或针对人群治疗时所获的利益和需注意的问题以及今后进一步研究的意义。 化学药物临床试验报告的结构与内容技术指导原则 二○○五年三月 已颁布
ICH E3 STRUCTURE AND CONTENT OF CLINICAL STUDY REPORTS 13 ICH E3 STRUCTURE AND CONTENT OF CLINICAL STUDY REPORTS 13. DISCUSSION AND OVERALL CONCLUSIONS “The discussion and conclusions should clearly identify any new or unexpected findings, comment on their significance and discuss any potential problems such as inconsistencies between related measures. The clinical relevance and importance of the results should also be discussed in the light of other existing data. Any specific benefits or special precautions required for individual subjects or at-risk groups and any implications for the conduct of future studies should be identified. Alternatively, such discussions may be reserved for summaries of safety and efficacy referring to the entire dossier (integrated summaries). ”
感谢您的参与,欢迎提问!