Download presentation
1
单基因遗传病与生化遗传 Single Gene Disorder (2)
井冈山大学医学院 生物学与医学遗传学教研室

2
先天性代谢缺陷(inborn errors of metabolism)
基因突变所引起的酶的结构改变或合成障碍,都有可能引起某种代谢过程的中断或紊乱。如果这种基因突变恰好发生在生殖细胞或受精卵中,就有可能传递给后代,从而使后代产生相应的先天性代谢缺陷(或遗传性酶病(hereditary enzymopathy)。 首先提出“先天性代谢缺陷”概念的Garrod
。 首先提出 先天性代谢缺陷 概念的Garrod.")
3
先天性代谢缺陷的共同规律 酶缺陷与酶活性 底物堆积和产物缺乏 底物分子的大小与性质 临床表型与酶缺陷

4
先天性代谢缺陷疾病的类型 糖代谢遗传病 氨基酸代谢遗传病 核酸代谢遗传病 脂类代谢遗传病 维生素代谢遗传病 金属代谢遗传病 药物代谢遗传病
……

5
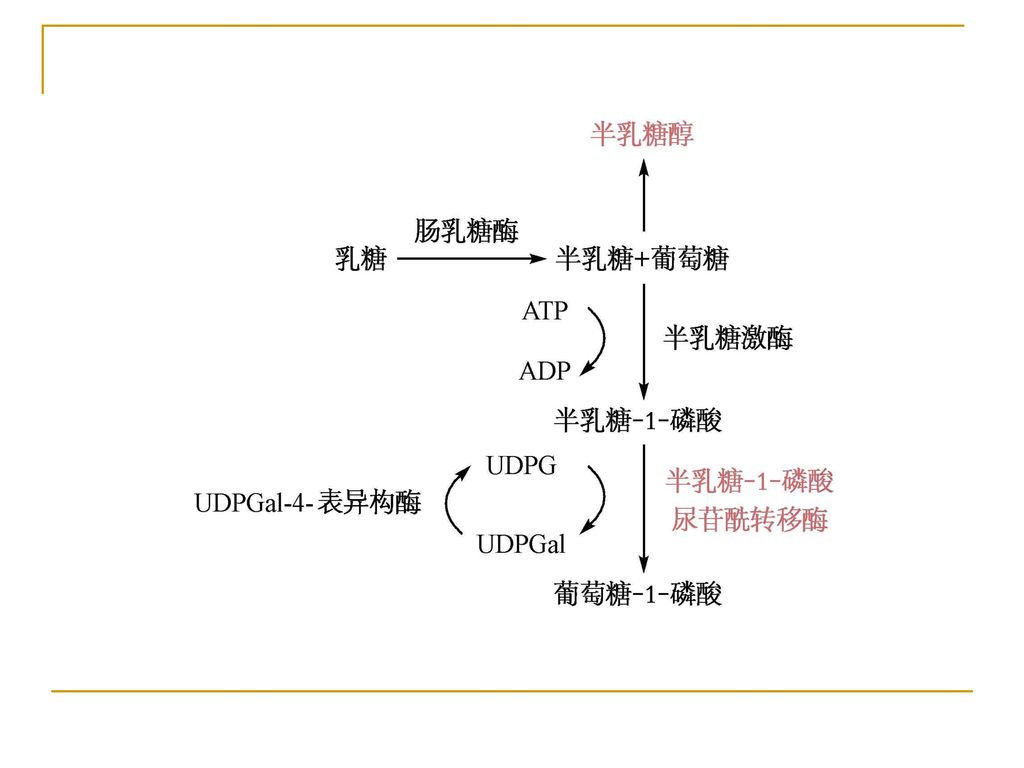
一、糖代谢障碍 半乳糖血症(galactosemia) 遗传学 半乳糖-1-磷酸尿苷酰转移酶(GPUT)缺陷,本病为常染色体隐性遗传,致病基因定位于9p13,发病率约为1/50000 。
 遗传学 半乳糖-1-磷酸尿苷酰转移酶(GPUT)缺陷,本病为常染色体隐性遗传,致病基因定位于9p13,发病率约为1/50000 。")
7
半乳糖和1-磷酸半乳糖在血中累积,部分随尿排出。1-磷酸半乳糖在脑、肾中累积可分别导致智力障碍、肝损伤甚至肝硬化及肾功能损伤等。
发病机制 半乳糖和1-磷酸半乳糖在血中累积,部分随尿排出。1-磷酸半乳糖在脑、肾中累积可分别导致智力障碍、肝损伤甚至肝硬化及肾功能损伤等。

8
临床表现 主要表现为患儿对乳糖不耐受,婴儿哺乳后呕吐、腹泻,继而出现白内障、肝硬化、黄疸、腹水、智力发育不全等。

9
半乳糖血症
11
诊断 实验室检查 测定酶活性 血和尿中半乳糖浓度 红细胞中半乳糖-1-磷酸的含量

12
预防及治疗 确诊后应立即停乳,代之豆浆。严重者应逐日监测尿内半乳糖水平。 新生儿筛查可早期诊断。可用羊水细胞或绒毛细胞培养物的酶学分析行产前诊断。

13
蚕豆病(葡萄糖-6-磷酸脱氢酶缺乏症) 遗传学 X连锁不完全显性,基因定位于Xq28,G6PD缺陷。 分布具有世界性,我国主要分布在长江以南,尤其是广东。
 遗传学 X连锁不完全显性,基因定位于Xq28,G6PD缺陷。 分布具有世界性,我国主要分布在长江以南,尤其是广东。")
14
6-p-glucose GSH G6PD 发病机制
G6PD缺陷,导致GSH生成减少,细胞抗氧化损伤能力下降,引起红细胞膜损伤;血红蛋白β链93位半胱氨酸巯基氧化,四聚体解离,形成Heinz小体,红细胞变形能力下降,破坏出现溶血。 蚕豆、伯氨喹啉等富含氧化物,服用后可加重该病。

15
临床表现 常见于10岁以下小儿,1~5岁为发病高峰。进食蚕豆后,蚕豆中的物质可使患儿体内的葡萄糖六磷酸脱氢酶被分解,导致大量红细胞破裂溶血,使患儿出现溶血性贫血。

16
发病轻重则与吃蚕豆多少无关,一般在进食蚕豆后1~2天内发病,早期病人常出现全身不适、胃口不佳、发热、头昏等酷似肝炎的症状。重症病人会由于大量红细胞被破坏而释放出胆红素,导致黄疸、浓茶样的血红蛋白尿、肝脾肿大,并有恶心呕吐、腹痛等临床症状,若不及时抢救治疗,发病后1 ~ 2天内就会死亡。

17
糖原贮积症 (glycogen storage disease,GSD)
遗传学 糖原贮积症是一类较罕见的遗传代谢病。由于酶的缺陷,使糖原在肝脏及肌肉中的代谢缺陷所致。根据所缺的酶不同,可将糖原贮积症分为Ⅰ~Ⅷ型,多数为常染色体隐性遗传,以Ⅰ型为最常见。

18
糖原贮积症的几种类型 病 名 OMIM 缺陷的酶 基因定位 症 状 GSD 0 240600 肝糖原合酶 AR,12p12.2 GSD Ⅰa
病 名 OMIM 缺陷的酶 基因定位 症 状 GSD 0 240600 肝糖原合酶 AR,12p12.2 GSD Ⅰa 232200 葡萄糖-6-磷酸酶 AR, 17q21 低血糖血症 GSD Ⅰb 232220 微体葡萄糖-6-磷酸转运 AR, 11q23 巨舌,肌张力减退 GSD Ⅰc 232240 微体磷酸吡咯转运 GSD Ⅱ 232300 α-1,4-葡糖苷酶 AR, 17q25.2 GSD Ⅱb 300257 XR, Xq24 GSD Ⅲ 232400 淀粉-1,6-葡糖苷酶 AR, 1p21 与I型相似 GSD Ⅳ 232500 淀粉-(1,4;1,6)转葡糖苷酶 AR, 3p12 肝脾肿大,肝硬化 GSD Ⅴ 232600 肌磷酸化酶 AR, 11q13 肌无力,肌痉挛 GSD Ⅵ 232700 肝磷酸化酶 AR, 14q21 低血糖症生长迟缓 GSD Ⅶ 232800 肌磷酸果糖激酶 AR, 12q13.3 肌痉挛肌无力肌痛 GSD Ⅷ 306000 磷酸化酶激酶 XR, Xp22.2 轻型低血糖症白内障 GSD Ⅸc 604549 AR
转葡糖苷酶. AR, 3p12. 肝脾肿大,肝硬化. GSD Ⅴ 肌磷酸化酶. AR, 11q13. 肌无力,肌痉挛. GSD Ⅵ 肝磷酸化酶. AR, 14q21. 低血糖症生长迟缓. GSD Ⅶ 肌磷酸果糖激酶. AR, 12q13.3. 肌痉挛肌无力肌痛. GSD Ⅷ 磷酸化酶激酶. XR, Xp22.2. 轻型低血糖症白内障. GSD Ⅸc AR.")
19
Ⅰ型糖原贮积症: 发病机制 葡萄糖-6-磷酸酶的基因缺陷,使肝、肾、及肠粘膜等组织中糖原蓄积。 临床表现 患者易出现低血糖,并有肝、肾肿大等症状,严重时会发生酸中毒。

20
基因定位于17q25.2,溶酶体内α-葡萄糖苷酶的缺乏,使糖原处理障碍,造成溶酶体内糖原堆积, 病变累及全身肌肉。
Ⅱ型糖原贮积症: 发病机制 基因定位于17q25.2,溶酶体内α-葡萄糖苷酶的缺乏,使糖原处理障碍,造成溶酶体内糖原堆积, 病变累及全身肌肉。 临床表现 一般在儿童期即发病,患者因心肌无力、心脏扩大而最终死于心力衰竭。

21
Ⅱ型糖原贮积症引起心脏扩大
22
粘多糖贮积症(mucopolysaccharidosis,MPS)
发病机制 粘多糖由结缔组织合成,是二糖重复单位串联而成的多糖链,粘多糖分解时需要多种酶的参与,这些酶的遗传性缺陷可导致粘多糖降解受阻,蓄积于溶酶体中形成粘多糖贮积症。

23
遗传学 粘多糖贮积症的几种类型 综合症名 缺 陷 的 酶 基因 遗传 主要症状 MPSⅠ α-艾杜糖醛酸酶 4p16.3 AR MPSⅡ
缺 陷 的 酶 基因 遗传 主要症状 MPSⅠ α-艾杜糖醛酸酶 4p16.3 AR MPSⅡ 磺艾杜糖醛酸硫酸酯酶 Xq28 XR 智力低下,肝脾肿大,骨骼异常 MSPⅢA 硫酸乙酰肝素硫酸酯酶 17q25.3 神经紊乱肝脾肿大 MSPⅢB N-乙酰α-氨基葡糖苷酸 17q21 MSPⅢC 14 MSPⅢD 12q14 MPSⅣA 硫酸软骨素硫酸酯酶 16q24.3 发育迟缓, 骨骼异常 MPSⅣB MPSⅤ β-半乳糖苷酶 3p21.33 与Hurler相似 MPSⅥ 软骨素-4-硫酸酯酶 5q11 与Huarler相似但症状 MPSⅦ β-葡糖苷酸酶 7q21-11 与Sanfilippl类似

24
患儿出现肝脾肿大、骨骼异常、智力障碍等症状,蓄积的粘多糖可随患儿的尿液排除。
临床表现 患儿出现肝脾肿大、骨骼异常、智力障碍等症状,蓄积的粘多糖可随患儿的尿液排除。 患儿骨骼异常 粘多糖贮积症患儿
26
常染色体隐性遗传性氨基酸代谢病,疾病基因已定位于12q24.1。
二、氨基酸代谢障碍 苯丙酮尿症(Phenylketonuria,PKU) 遗传学 常染色体隐性遗传性氨基酸代谢病,疾病基因已定位于12q24.1。 国外发病率约1/4500~1/100000,我国发病率约为1/16500。
 遗传学. 常染色体隐性遗传性氨基酸代谢病,疾病基因已定位于12q24.1。 国外发病率约1/4500~1/100000,我国发病率约为1/16500。")
27
发病机制 PKU患者PAH基因突变使患者肝脏内PAH缺乏,苯丙氨酸不能转变为酪氨酸,后者转化为苯丙酮酸和苯乳酸并在体内累积,并导致血液和尿液中苯丙氨酸及其衍生物排出增多。

29
临床表现 临床上表现为精神发育迟缓,皮肤、毛发和虹膜色素减退,头发呈赤褐色,癫痫,湿疹,特殊的鼠样臭味尿。患儿在出生后若不及早得到低苯丙氨酸饮食治疗,便出现不可逆的大脑损害和严重的智力发育障碍。

30
PKU患者精神发育迟缓,皮肤、毛发和虹膜色素减退,头发呈赤褐色
31
诊断 临床表现 新生儿筛查 尿三氯化铁试验 血氨基酸分析 酶学分析 DNA分析

32
PKU的分子诊断

33
治疗 目前临床上常在婴儿出生后立即进行PKU的筛查,一经肯定,立即给患儿停乳,喂给低苯丙氨酸水解蛋白,禁荤食、乳类、豆类和豆制品,可以达到临床痊愈。

34
右1为PKU患者;左1为其妹,经产前诊断后确诊亦为患者,出生后低苯丙氨酸饮食,达到临床痊愈。

35
白化病(albinism) 遗传学 发病机制 酪氨酸酶基因缺陷,遗传方式为AR;致病基因定位于11q14-q21。
患者体内酪氨酸酶酶缺乏,不能有效地催化酪氨酸转变为黑色素前体,最终导致代谢终产物黑色素缺乏。

36
酪氨酸酶

37
临床表现 全身白化;视网膜 无色素,畏光。 视网膜无色素 白化病患者
38
本病呈常染色体隐性遗传, 疾病基因定位于 3q21-q23,患者尿黑酸氧化酶缺陷。
尿黑酸尿症(alkaptonuria) 遗传学 本病呈常染色体隐性遗传, 疾病基因定位于 3q21-q23,患者尿黑酸氧化酶缺陷。
 遗传学. 本病呈常染色体隐性遗传, 疾病基因定位于 3q21-q23,患者尿黑酸氧化酶缺陷。")
39
发病机制 尿黑酸氧化酶

40
临床表现 病人的尿中含有尿黑酸(alkapton),曝光后可变为黑色的物质,这种病症在婴儿期就可表现出来,到成年时由于尿黑酸大量沉积于关节与软骨外,使关节变性。一般无明显的临床表现,严重时可出现关节炎,并发心脏病。
,曝光后可变为黑色的物质,这种病症在婴儿期就可表现出来,到成年时由于尿黑酸大量沉积于关节与软骨外,使关节变性。一般无明显的临床表现,严重时可出现关节炎,并发心脏病。")
42
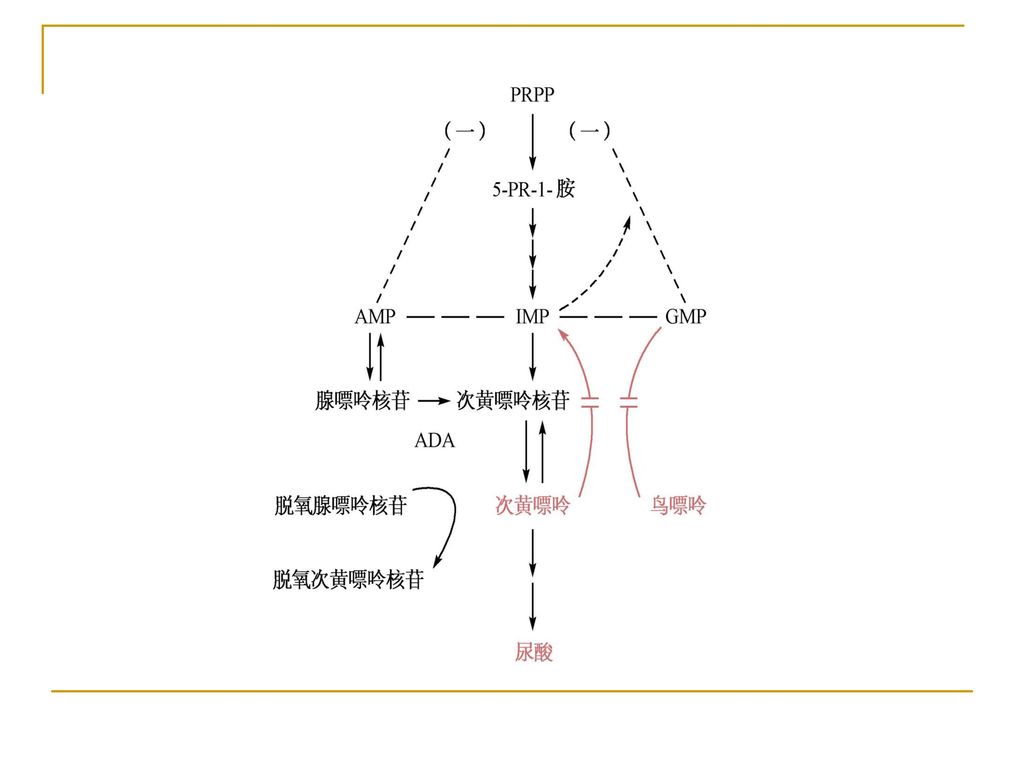
本病是一种由于次黄嘌呤鸟嘌呤磷酸核糖转移酶(HGPRT)缺陷所致的疾病,XR,基因定位于
三、核酸代谢障碍 次黄嘌呤鸟嘌呤磷酸核糖转移酶缺陷症 (Lesch-Nyhan sydrome) 遗传学 本病是一种由于次黄嘌呤鸟嘌呤磷酸核糖转移酶(HGPRT)缺陷所致的疾病,XR,基因定位于 Xp26-p27.2。
 遗传学. 本病是一种由于次黄嘌呤鸟嘌呤磷酸核糖转移酶(HGPRT)缺陷所致的疾病,XR,基因定位于. Xp26-p27.2。")
43
发病机制 HGPRT是体内核酸补救合成途径的关键酶,它的缺陷使黄嘌呤、鸟嘌呤向相应核苷酸的转化受阻,底物在体内堆积,特别是在神经系统中的堆积,进而引起发病。

45
临床表现 患者一般为男性,出生3~4个月开始出现神经系统症状,激惹不安,烦躁,运动发育迟缓;约1岁后出现舞蹈样手足徐动,肌张力高,下肢呈剪刀样交叉;约2 ~ 3岁起,表现强迫性自我摧残行为,多数智商低于65;患儿精神发育迟滞,强直性大脑性瘫痪。

47
红细胞、皮肤成纤维细胞测定HGPRT酶活性 测晨尿的尿酸/肌酐比值可作筛选手段 利用羊水细胞检查可行产前诊断

48
现尚无特殊治疗方法,一般采用口服黄嘌呤氧化酶的抑制剂—别嘌呤醇,可抑制尿酸的生成,防止尿酸结石和肾脏病损,但不改善神经系统症状。
饮食尽量无嘌呤摄入。

49
着色性干皮病 (xeroderma pigmentosum, XP)
遗传学 着色性干皮病为常染色体隐性遗传病。由于患者体内缺乏核酸内切酶引起的疾病。本病可分为(XPA-XPG)7型,目前已克隆出XPA、XPB、XPC、XPD的基因,其中XPA定位于9q34.1, XPB定位于2q21。
7型,目前已克隆出XPA、XPB、XPC、XPD的基因,其中XPA定位于9q34.1, XPB定位于2q21。")
50
核酸内切酶缺乏→不能切除由紫外线诱发的嘧啶二聚体(特别是胸腺嘧啶二聚体,T-T)。
发病机制 核酸内切酶缺乏→不能切除由紫外线诱发的嘧啶二聚体(特别是胸腺嘧啶二聚体,T-T)。
。")
52
临床表现 患者皮肤对阳光过敏,日照后可出现红斑、水肿、色素沉着、干燥、角化过度及萎缩等皮损。有些病人智能落后,感音神经性聋及共济失调。易患基底细胞癌、鳞癌、恶性黑色素瘤等,均伴有免疫系统的异常。

53
着色性干皮病
54
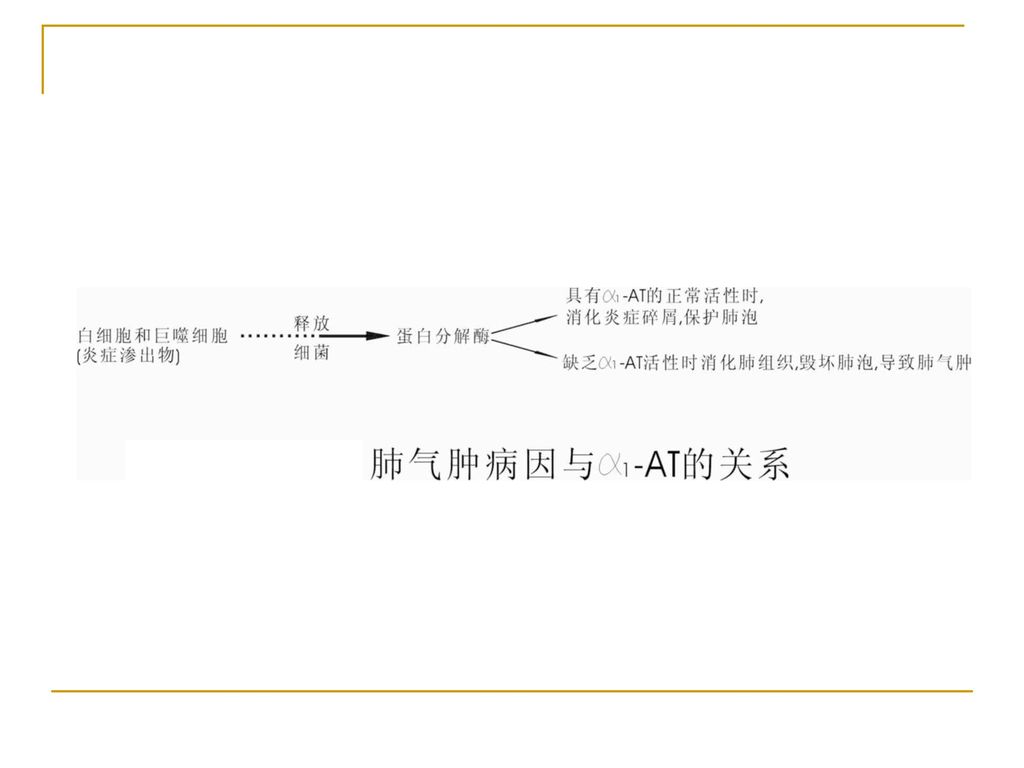
四、抗胰蛋白酶缺乏症 (α1-antitrypsin )
遗传学(AR) α1抗胰蛋白酶为糖蛋白 基因定位于14q32.1
 α1抗胰蛋白酶为糖蛋白. 基因定位于14q32.1.")
55
发病机制 α1抗胰蛋白酶存在于血浆中,尿液、支气管分泌物等,可抑制血清中多种蛋白酶活性。 α1抗胰蛋白酶缺乏症特征是血清中α1-AT水平下降。

57
本 章 节 重 点 分子病及遗传性酶病的概念; 镰状红细胞性贫血及发病机制; 地中海贫血及发病机制; 氨基酸代谢病的发病机理。

58
【复习思考题】 1、从分子遗传学角度说明镰形红细胞贫血及α地中海贫血、β地中海贫血发生的机制及特点。
2、苯丙酮尿症,白化病的发病机理分别是什么? 何为血红蛋白病?血红蛋白病如何分类?




. 免疫缺陷病 (Immunodeficiency diseade,IDD) : 由免疫系统中任何一个成分在发生、发 育和成熟过程中的缺失或功能不全而导致免 疫功能障碍所引起的疾病。 免疫缺陷病分为 : 先天性 />")







 3 、遗传咨询( 4 个步骤)、产前检查 4 、人类基因组计划.>")










