Download presentation
1
神经皮肤综合征 (斑痣性错构瘤病) 这是一组起源于外胚层结构的先天性畸形 共同特点:既有皮肤病变,又有明显的神经系统异常表现
 这是一组起源于外胚层结构的先天性畸形 共同特点:既有皮肤病变,又有明显的神经系统异常表现")
2
常见疾病 神经纤维瘤病 结节性硬化 脑颜面血管瘤综合征 血管母细胞瘤病

3
少见疾病 K-T综合征 共济失调性毛细血管扩张症 神经皮肤黑素沉着症 表皮痣综合征 基底细胞痣综合征 遗传性出血性毛细血管扩张症

4
神经纤维瘤病分型 可以分为两个亚型,即神经纤维瘤病I型和II型。
神经纤维瘤病I 型也叫von Recklinghausen病,以前也称周围型。II 型以前也称为中央型 由于I型除了皮肤特征性改变外,也常伴有颅内病变,II型虽以中枢神经系统肿瘤为主要表现,也可有周围皮肤异常表现,故现在基本不采用这种分类法。

5
神经纤维瘤病I型

6
诊断标准 由1988年美国国家健康协会制订 凡符合以下两条或两条以上表现者即可作出神经纤维瘤病I型的诊断: (1)6个或更多皮肤牛奶咖啡斑
(2)一个丛状神经纤维瘤或两个以上神经纤维瘤 (3)两个或更多色素沉着性虹膜错构瘤 (4)多发性腋部或腹股沟区雀斑 (5)视神经胶质瘤 (6)一级亲属患有神经纤维瘤病I型 (7)特征性骨病变如:蝶骨大翼发育不全、假关节、 先天性长骨弯曲、飘带状肋骨等。
一个丛状神经纤维瘤或两个以上神经纤维瘤. (3)两个或更多色素沉着性虹膜错构瘤. (4)多发性腋部或腹股沟区雀斑. (5)视神经胶质瘤. (6)一级亲属患有神经纤维瘤病I型. (7)特征性骨病变如:蝶骨大翼发育不全、假关节、 先天性长骨弯曲、飘带状肋骨等。")
7
病因病理学 它属于常染色体显性遗传性疾病,有关基因是在染色体17长臂。 50%有基因突变
神经纤维瘤病I型主要影响神经外胚层,同时也影响中胚层,他们既产生肿瘤性病变,又可产生发育不良性改变。

8
临床表现 神经纤维瘤病 I型约占整个神经纤维瘤病的90% 发病率占出生存活儿1:3000~5000,儿童多见。 很多人没有任何临床症状
绝大多数病人的诊断作出是由于病人有明显异常的皮肤改变,如牛奶咖啡斑腋部雀斑,皮肤神经纤维瘤等 。 由于蝶骨翼发育不良导致颞叶疝入眶内引起搏动性突眼

9
临床表现 视神经胶质瘤和青少年青光眼可引起视力下降 约30-45%儿童有学习能力低下 约半数病人因脑实质增加而表现头大
其他表现包括中胚层发育不良引起的脊柱侧后突、胫腓骨骨皮质变薄弯曲、假关节形成等。

10
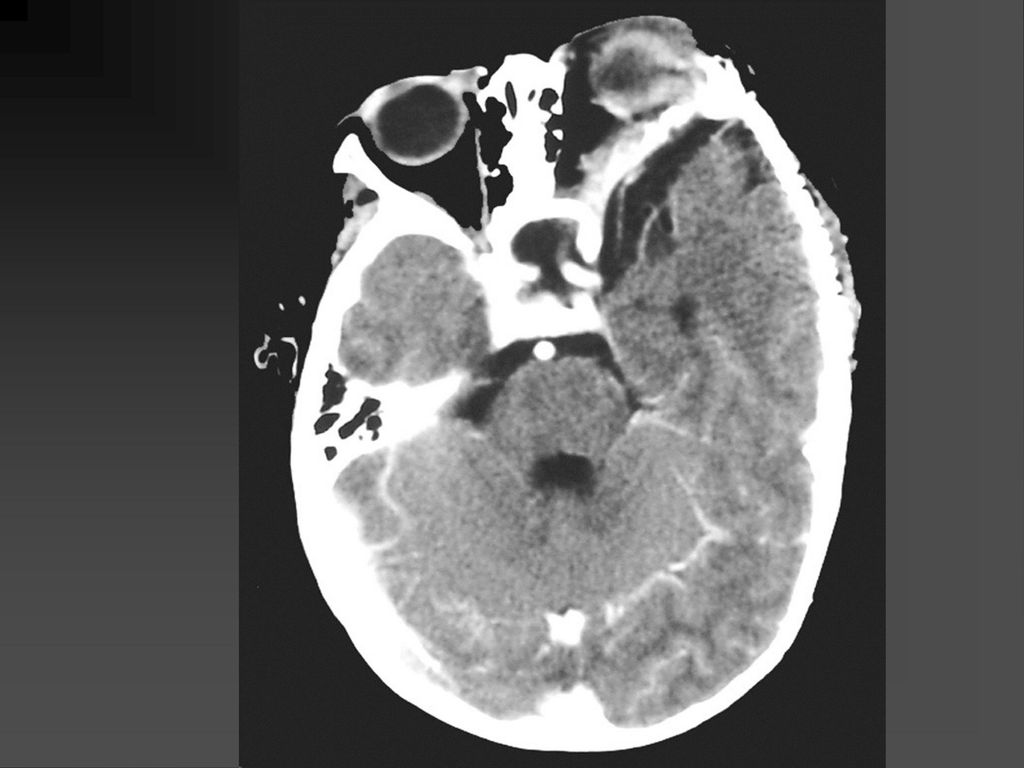
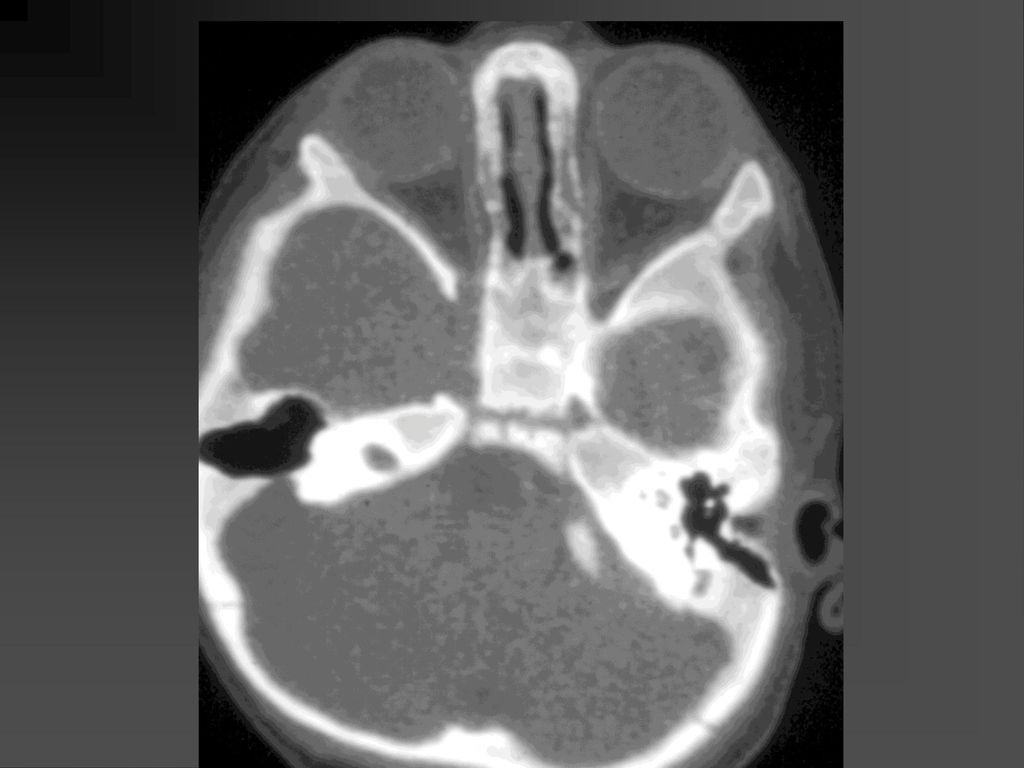
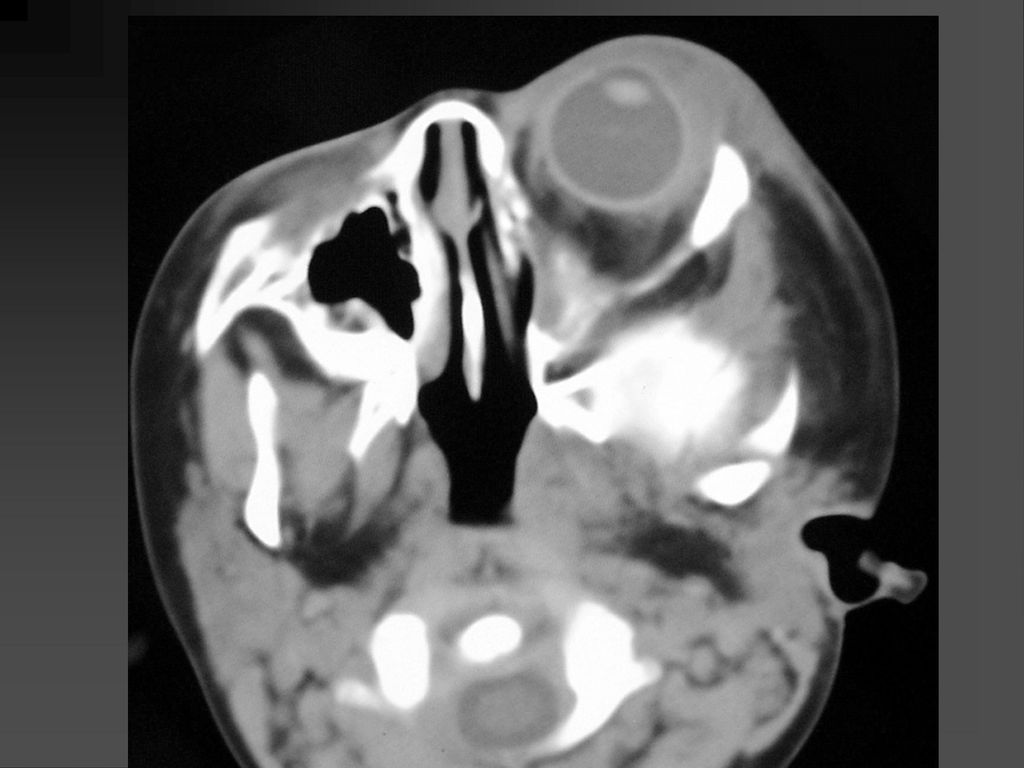
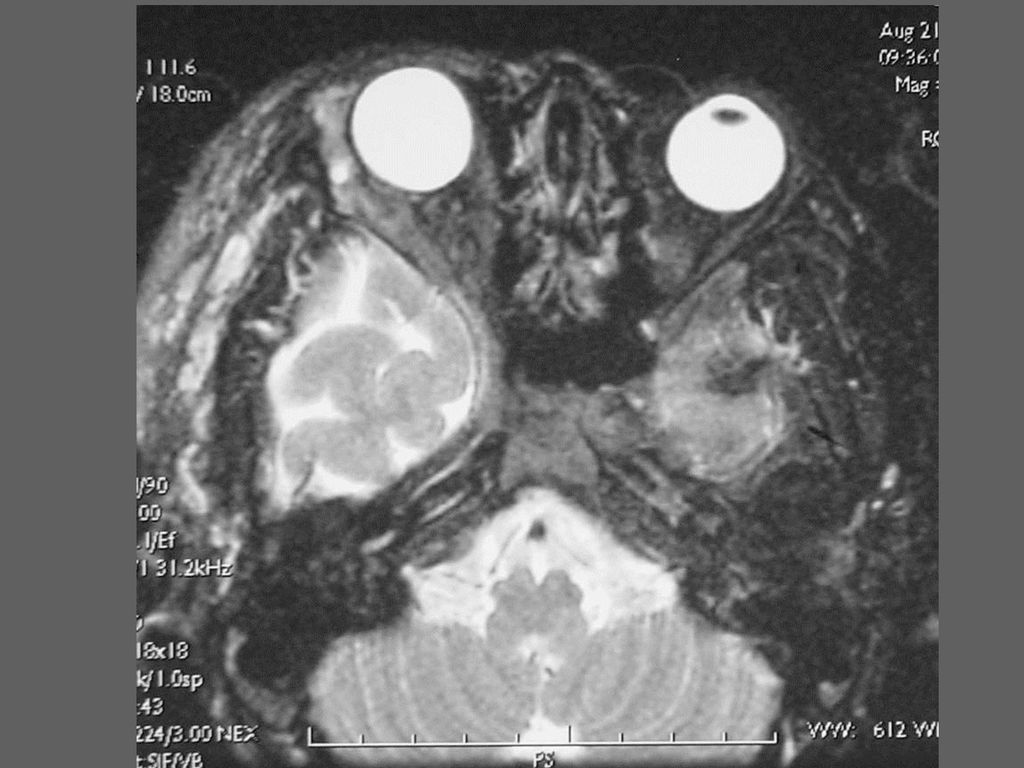
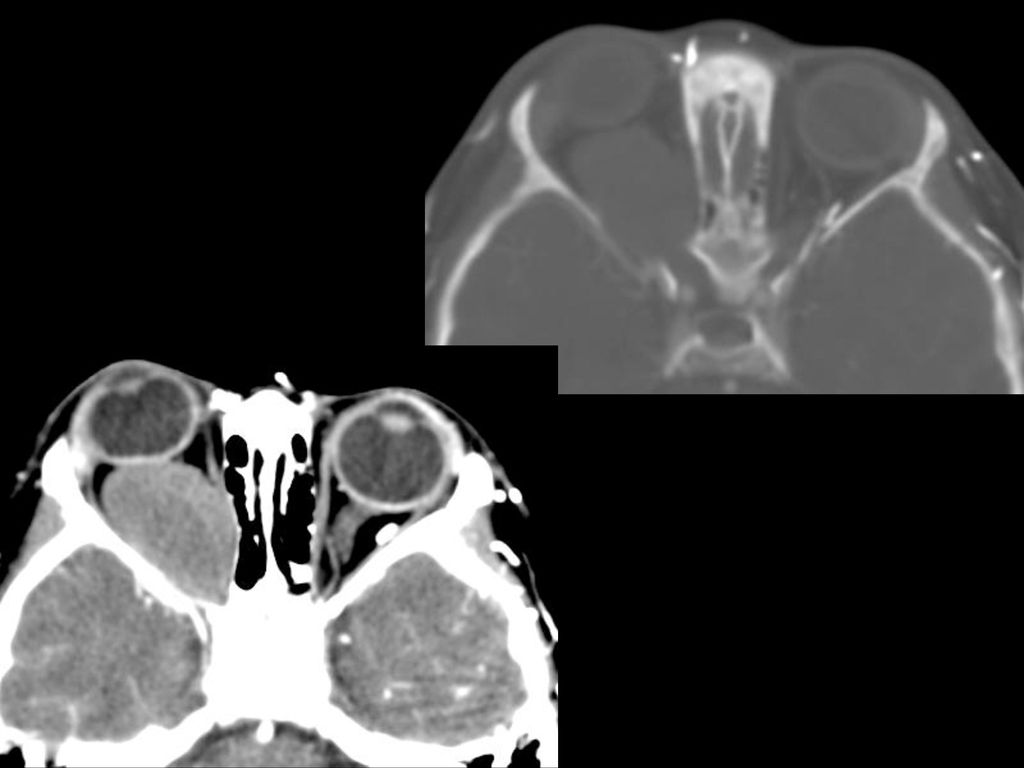
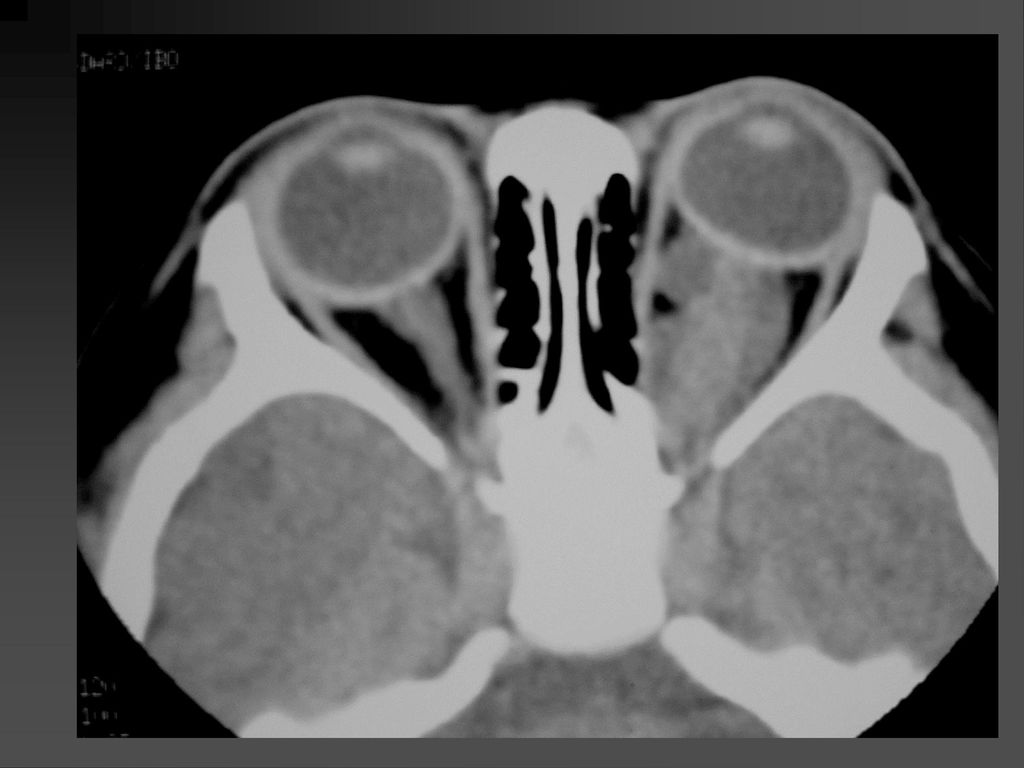
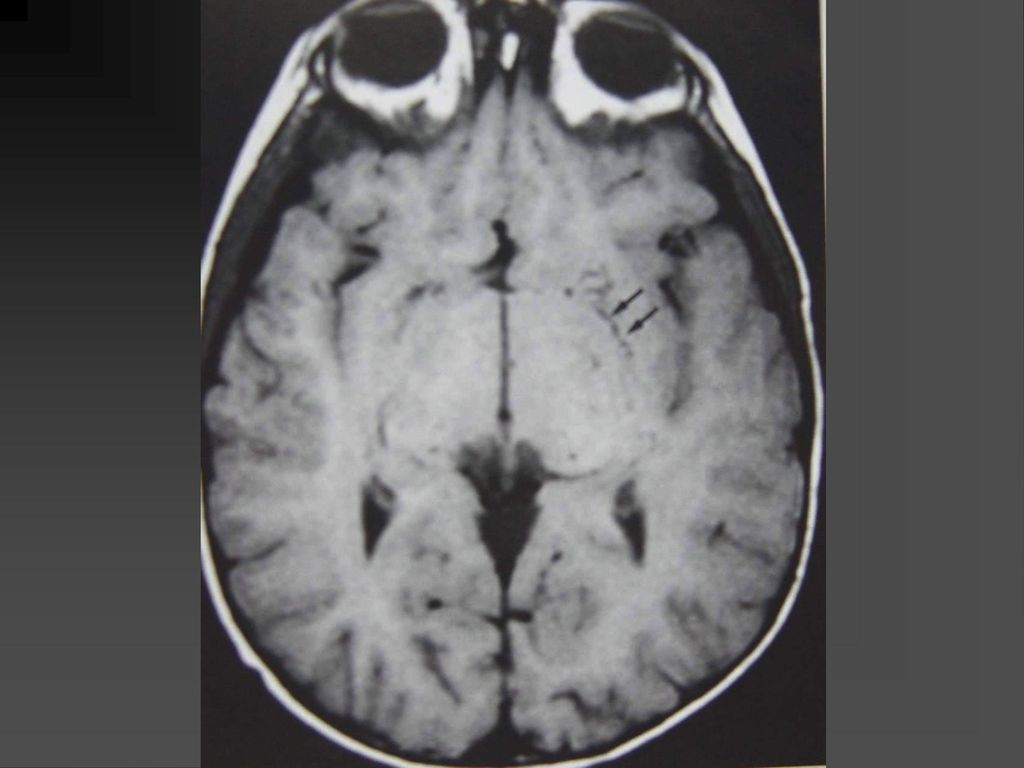
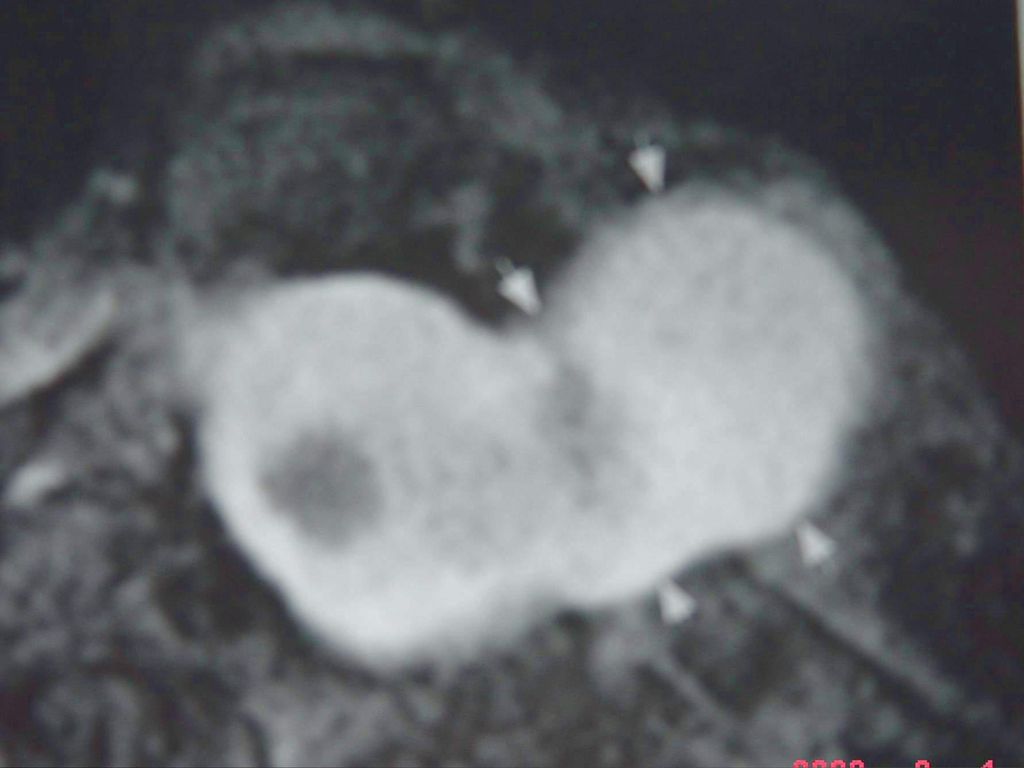
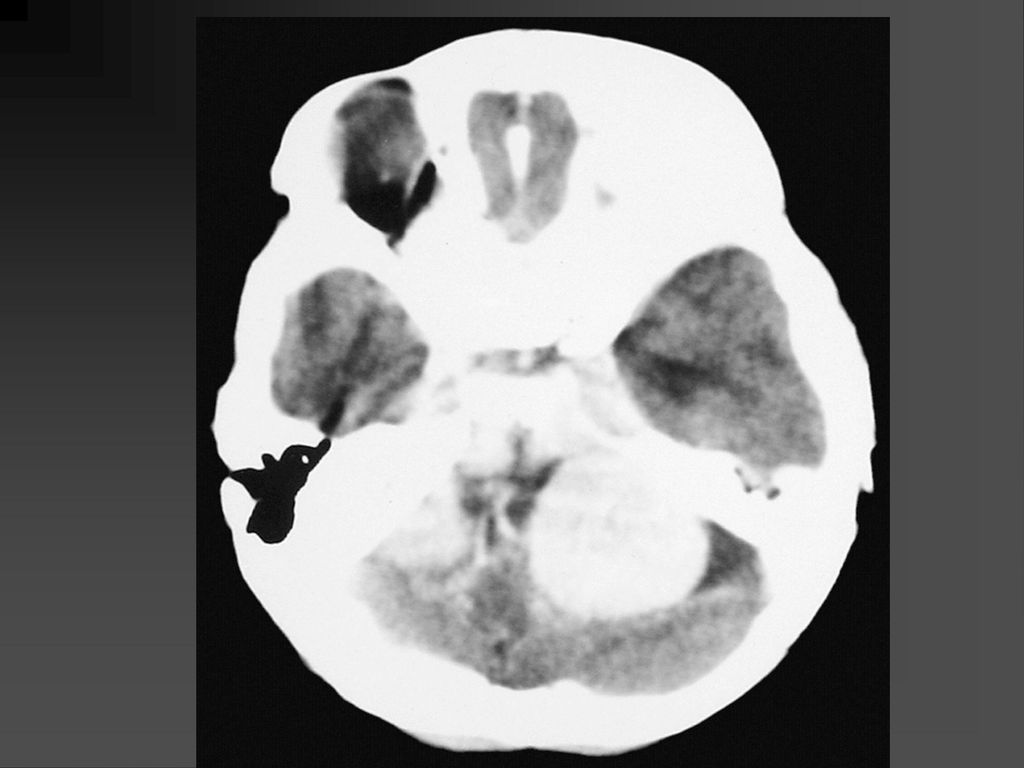
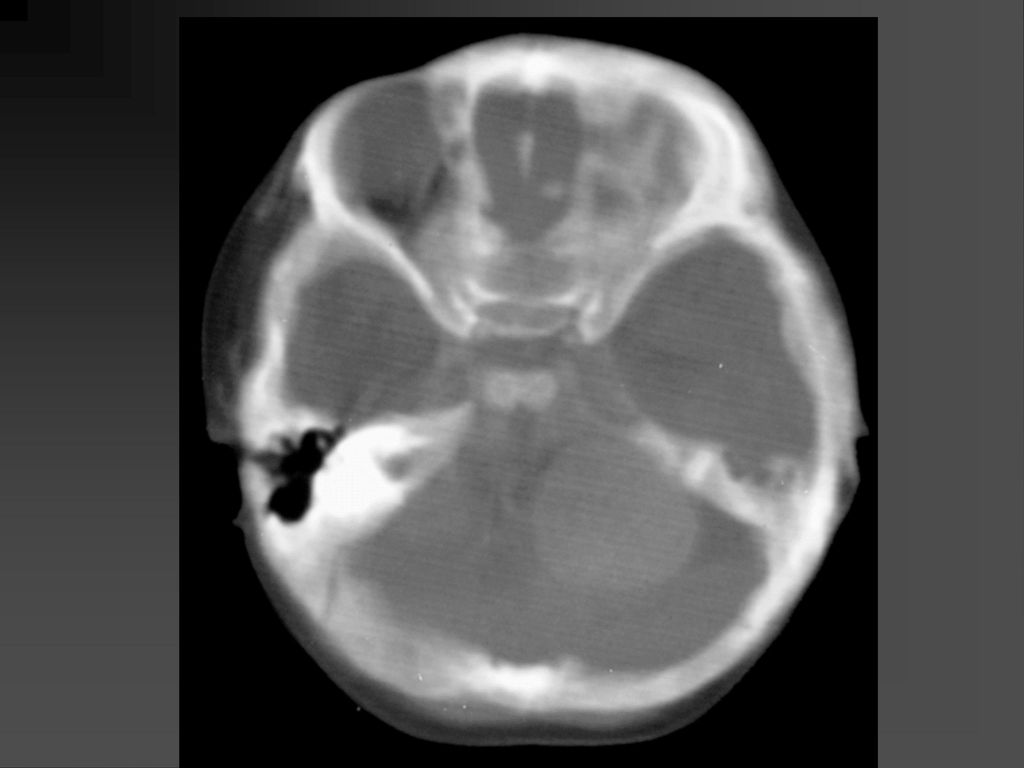
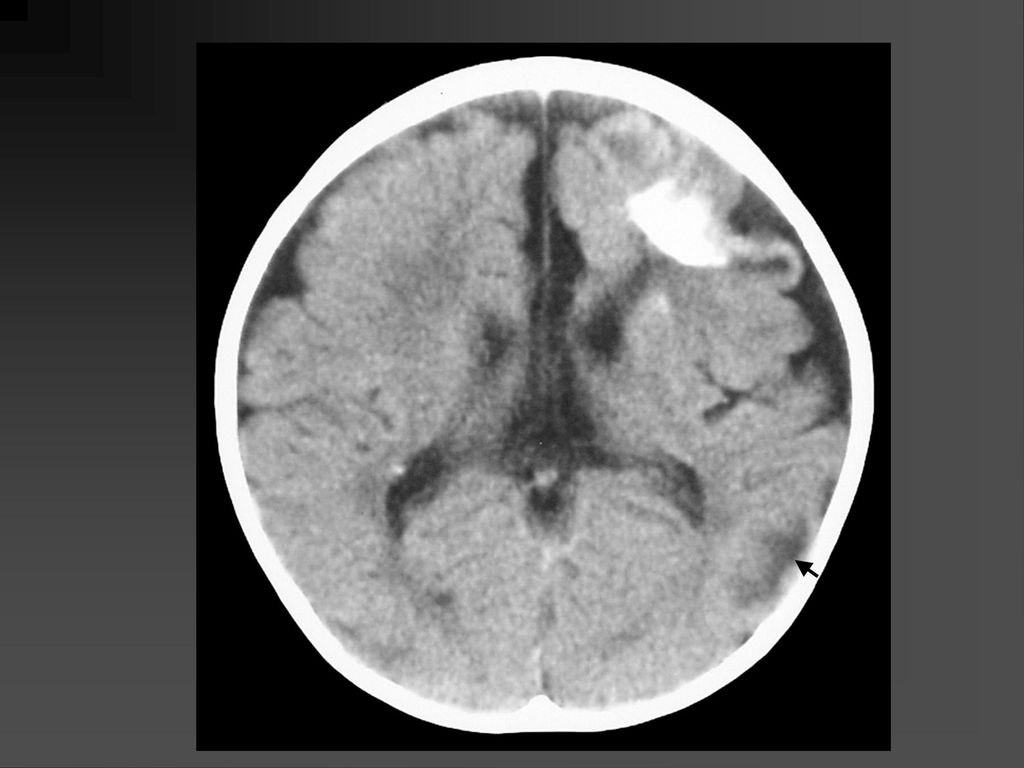
蝶骨翼发育不全、甚至蝶骨翼缺如,通常是单侧性的。
中颅凹底部的颞叶通过发育不全的蝶骨翼向前突入眼眶,使眼球外突。 蝶骨翼发育不全者常同时伴有邻近眼睑和眶内颞部丛状神经纤维瘤,肿瘤呈弥漫性生长。

15
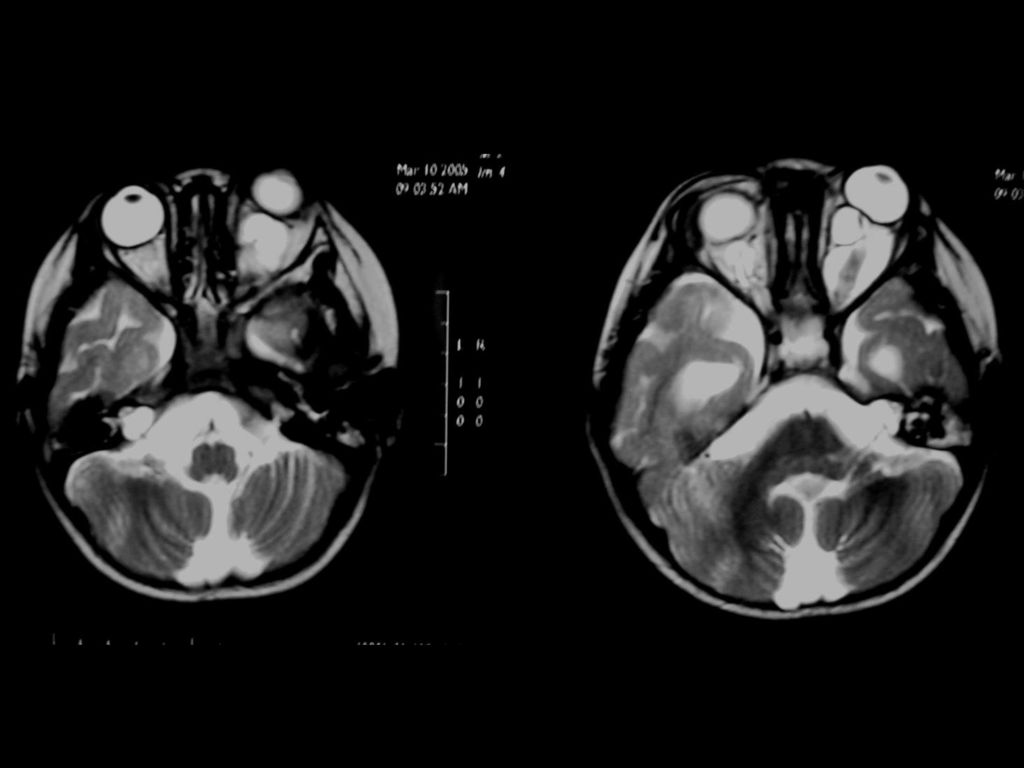
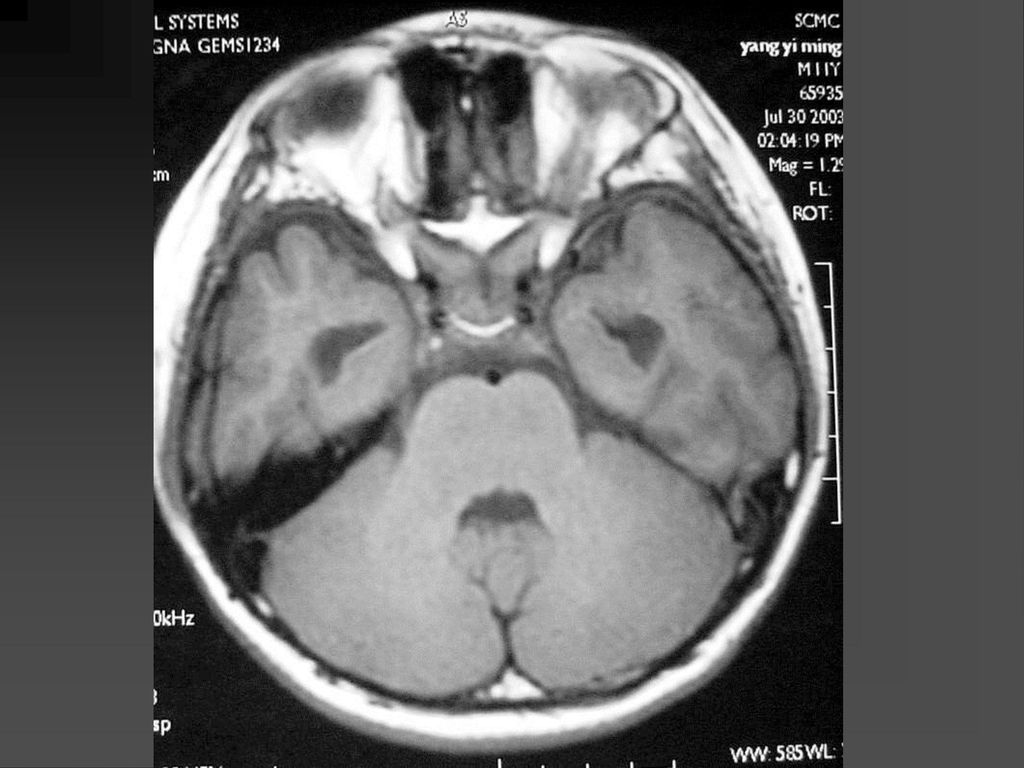
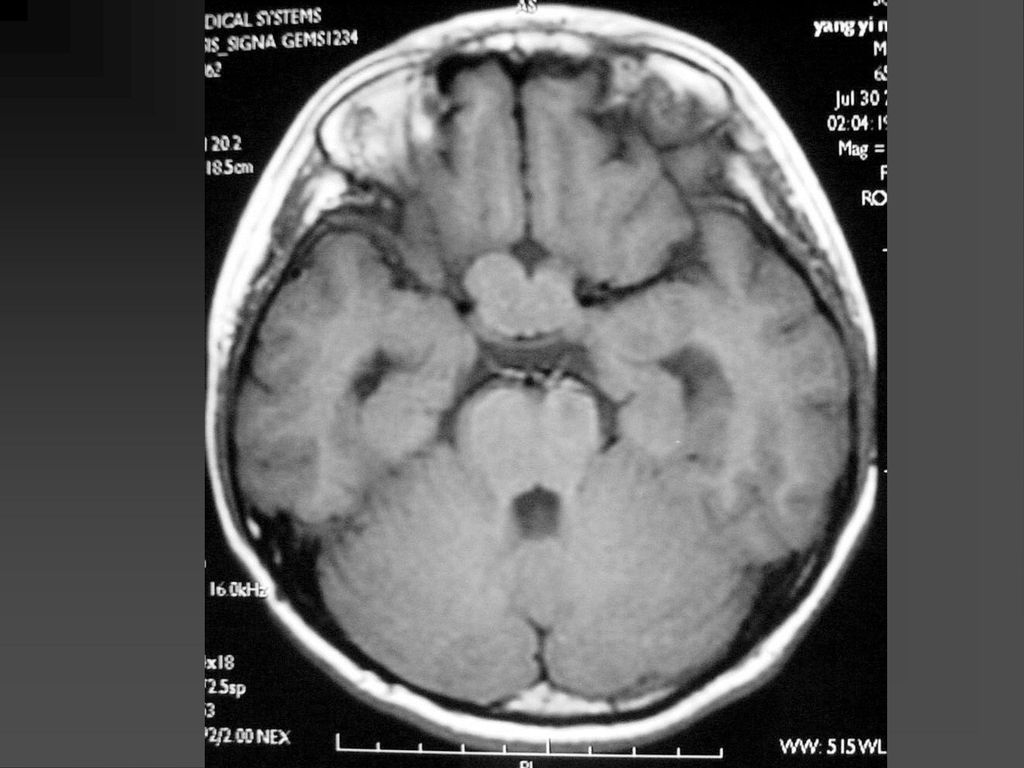
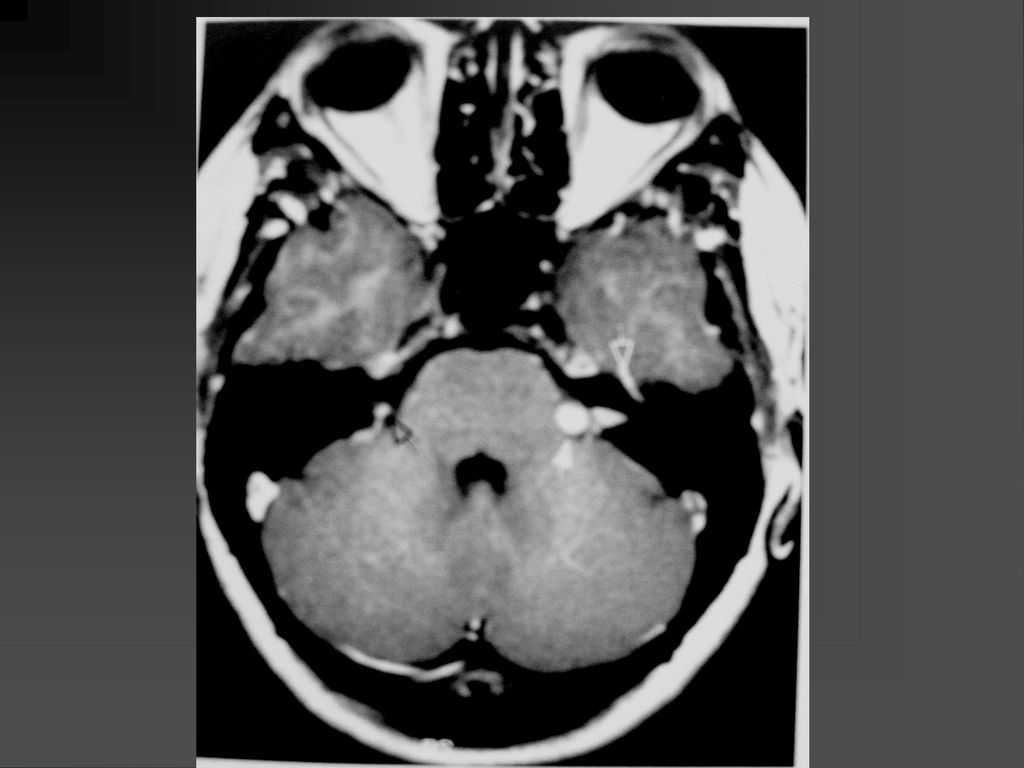
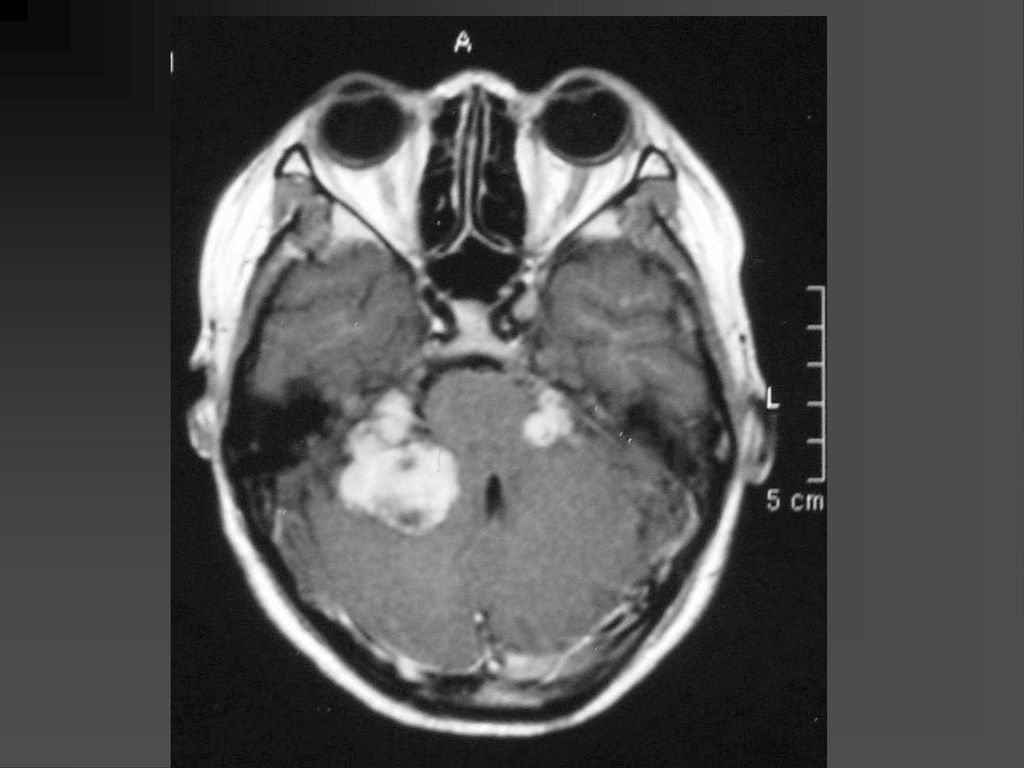
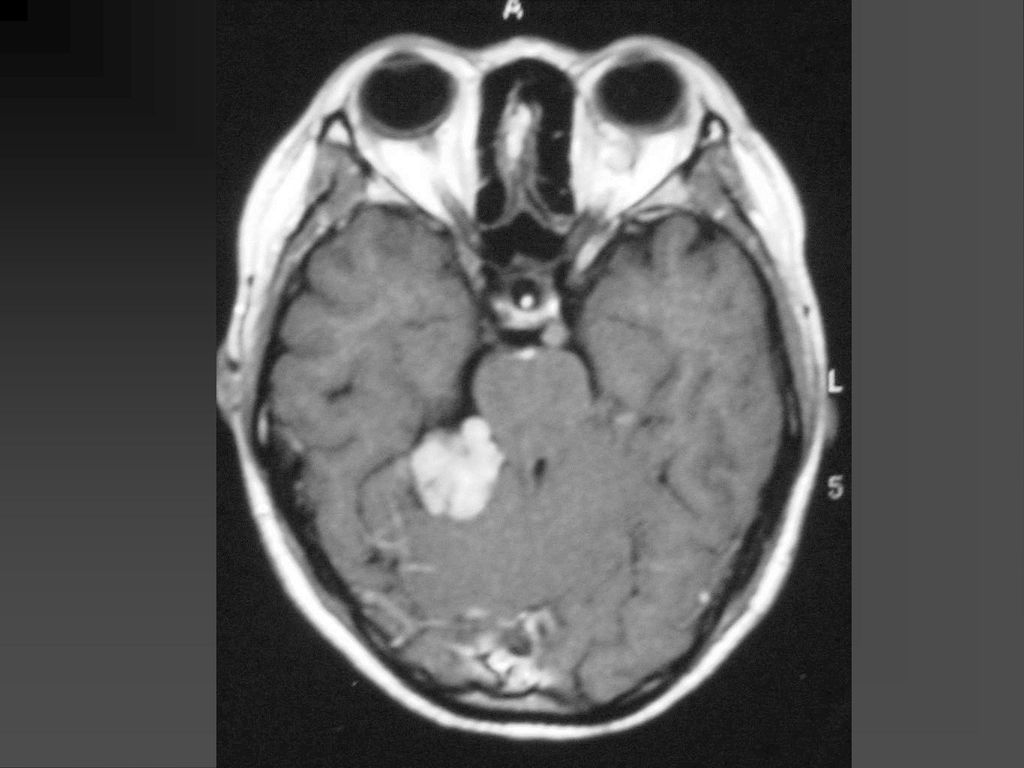
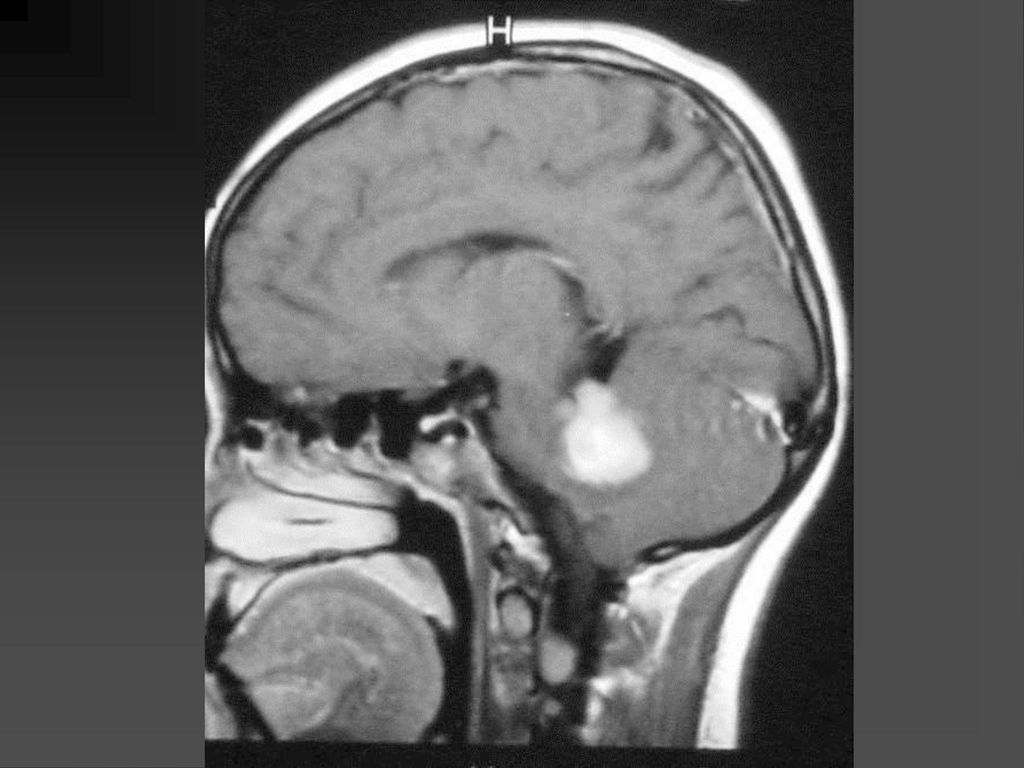
是神经纤维瘤病I型中最常见的肿瘤,约占10-15%
常是低级星形细胞瘤 真正产生临床症状者仅占其中半数 视神经胶质瘤可以是一侧性也可以两侧性,可以延伸至视交叉、视束。

26
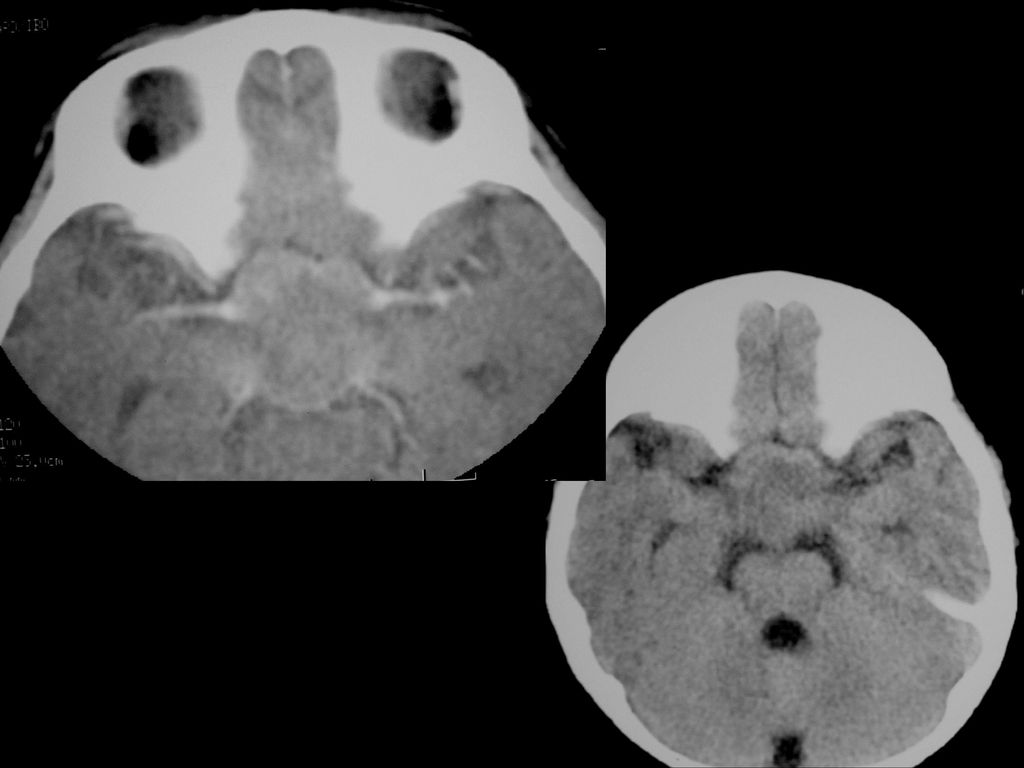
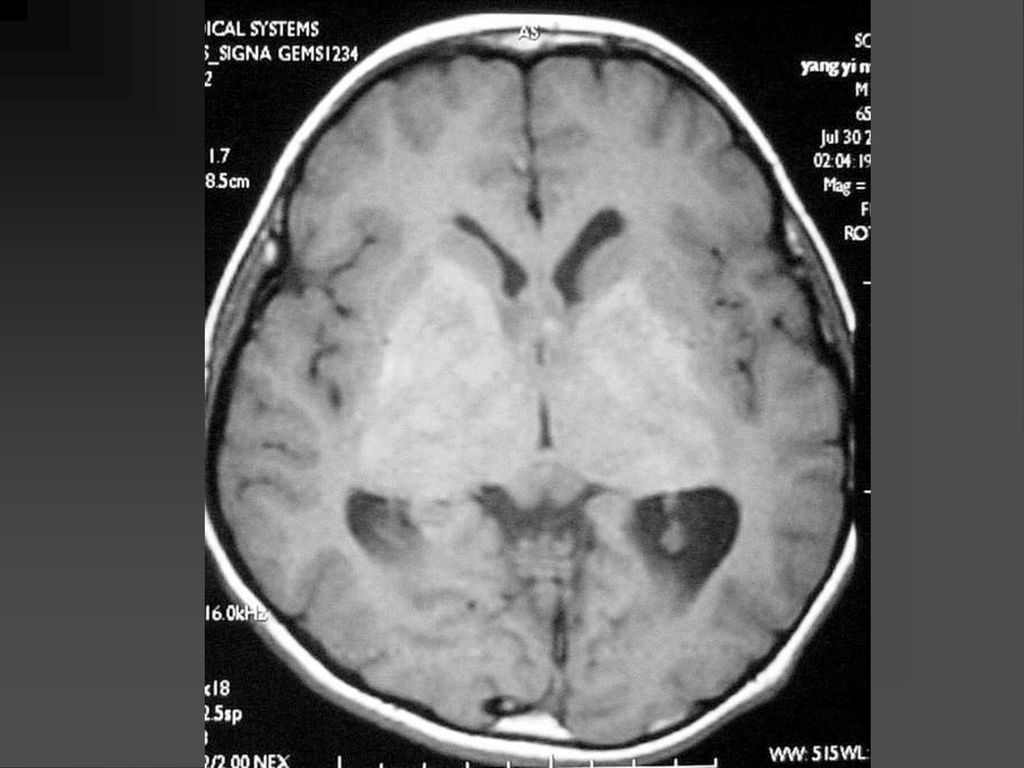
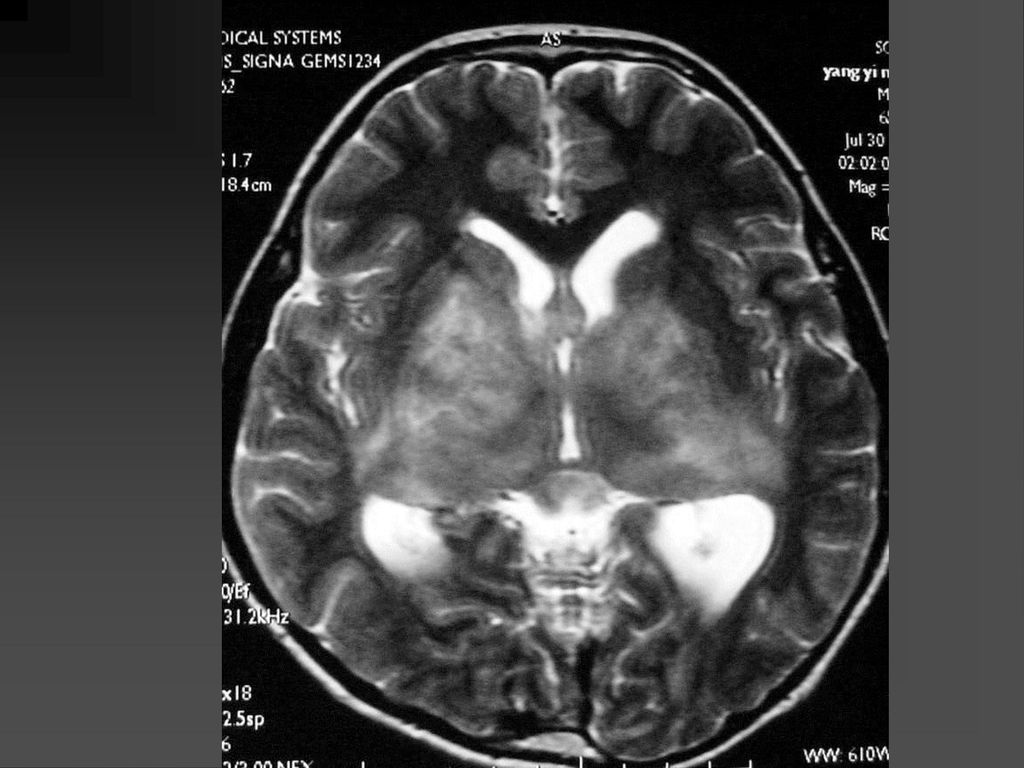
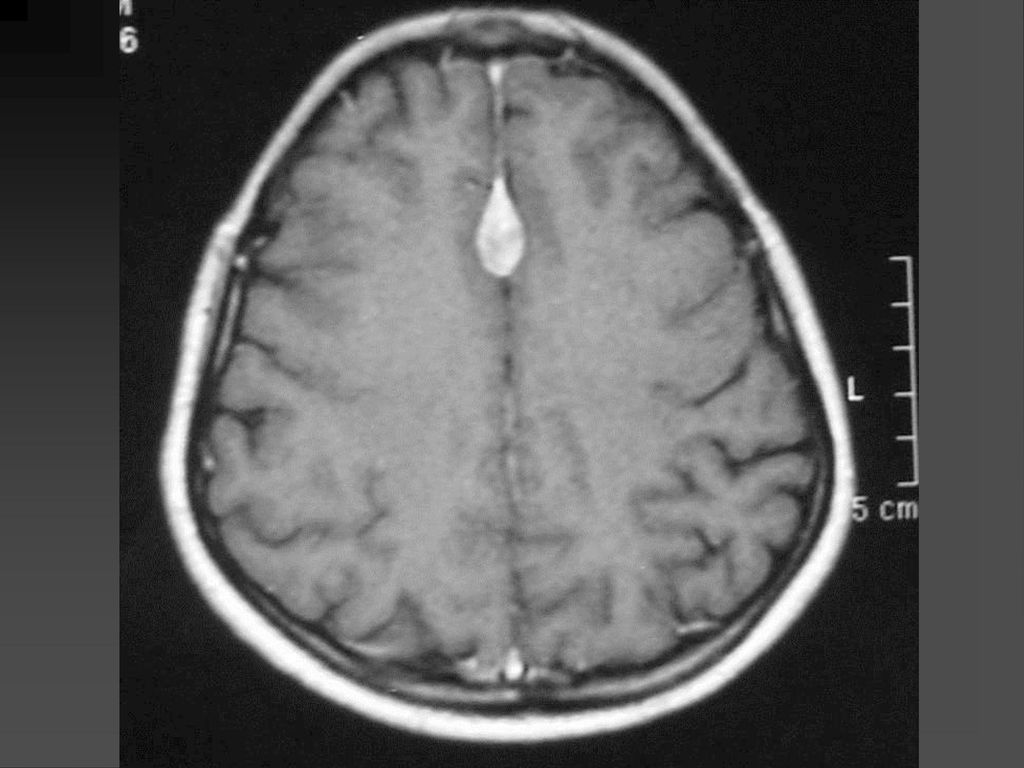
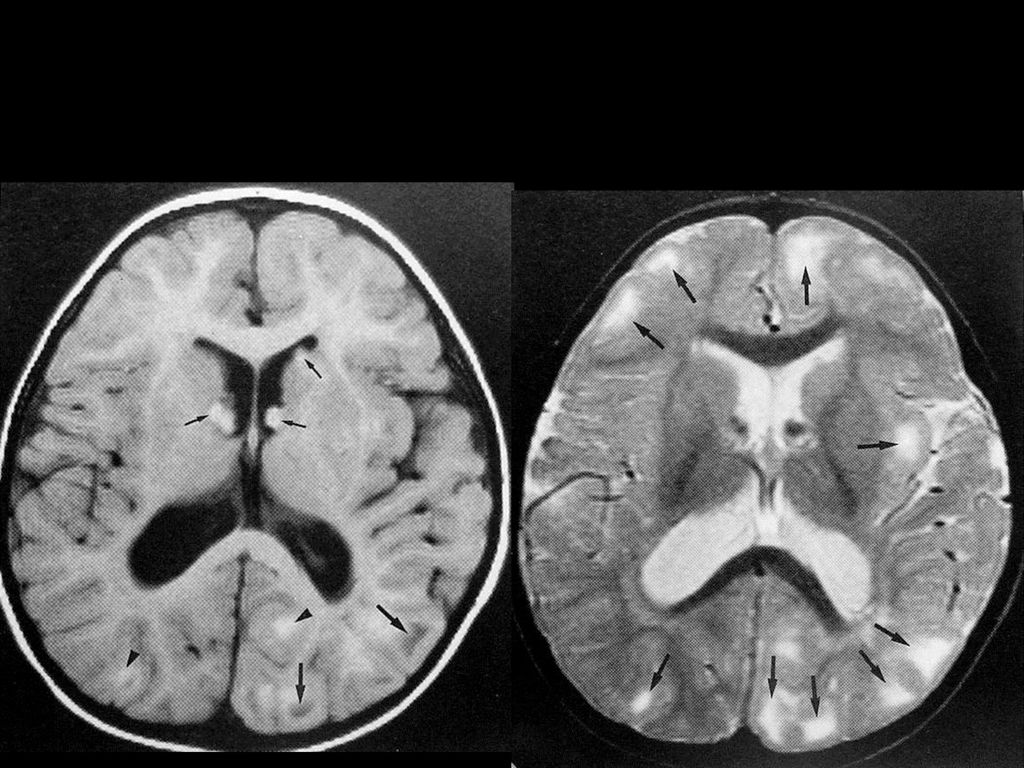
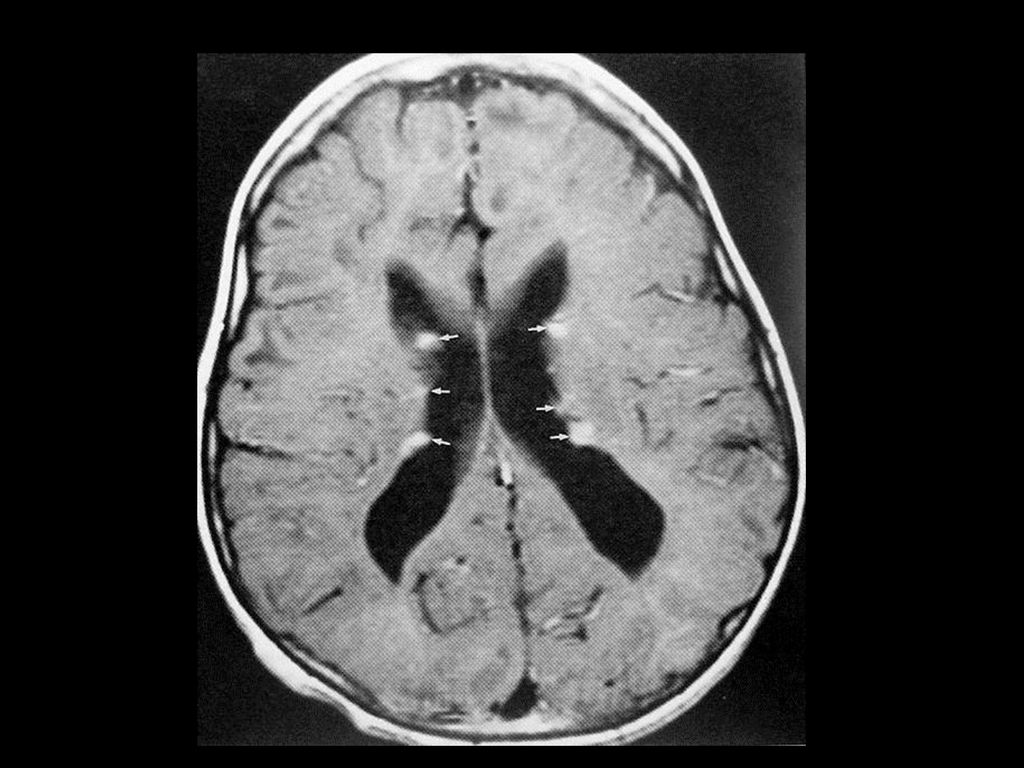
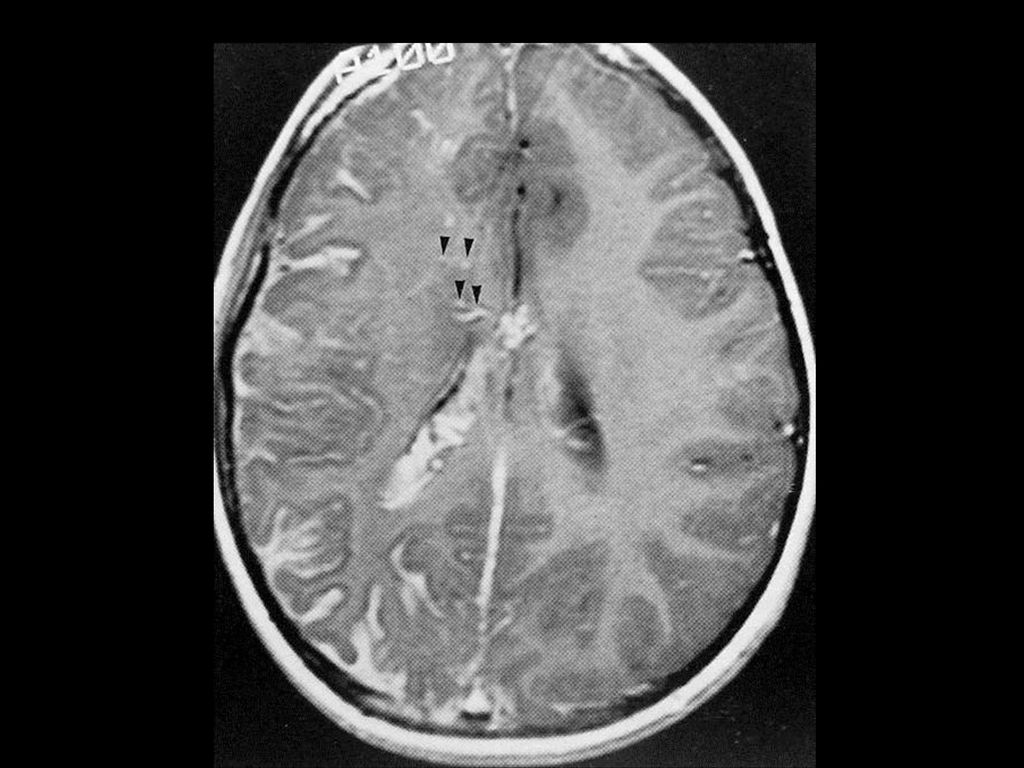
80%病人颅内有非肿瘤性的错构瘤病变或髓鞘空泡样变性
病变位于苍白球、丘脑、脑干、小脑白质和小脑齿状核等。 CT敏感性逊于MRI,绝大多数呈等密度,少数低密度。 若基底节病变出现明显占位效应且增强后有强化,可能提示病变向胶质瘤转化,需密切随访。

35
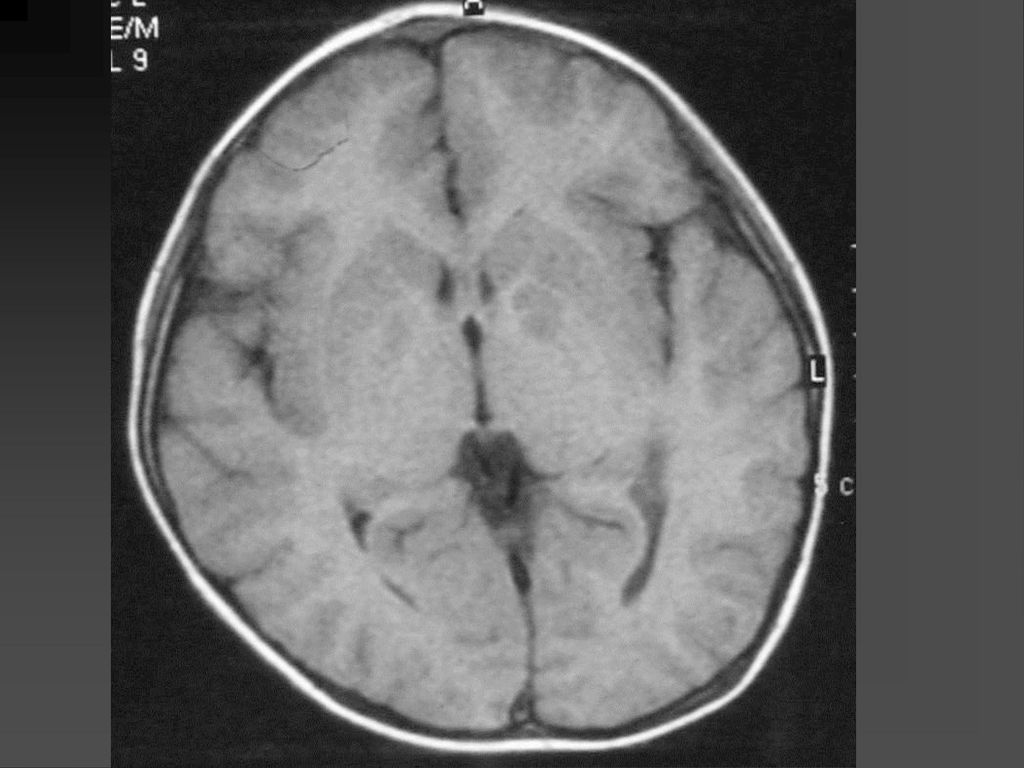
其他颅内表现 脑积水 颈内动脉或大脑前、中动脉起始部狭窄,有时可显示由此引起的基底节脑缺血改变等。

38
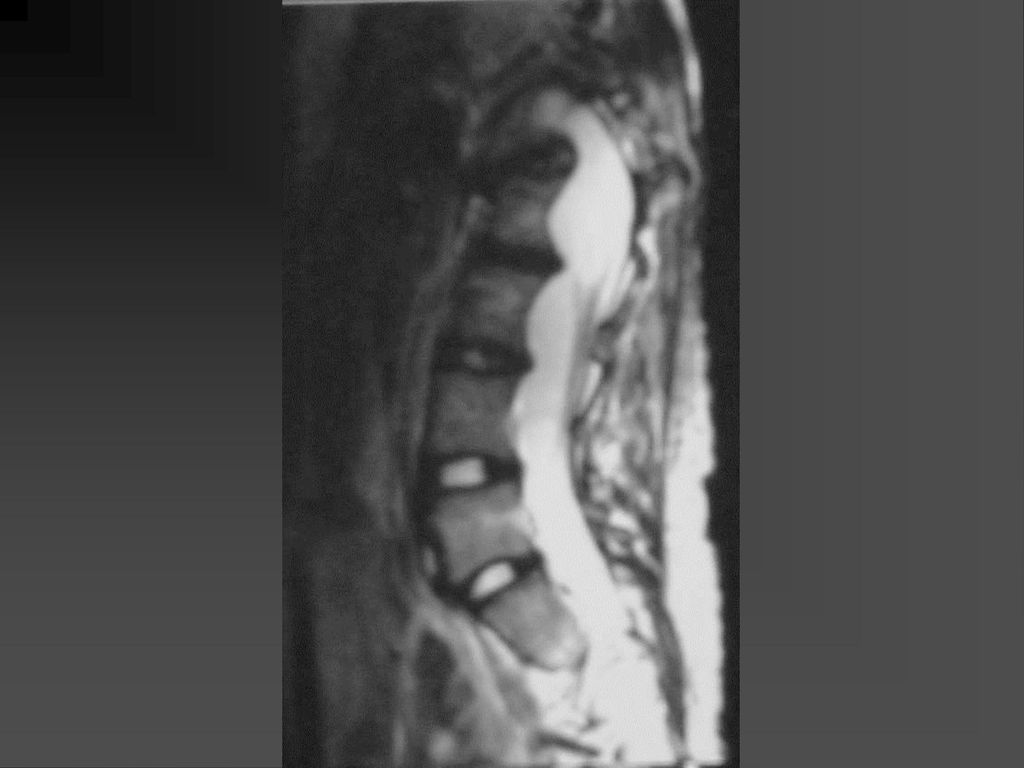
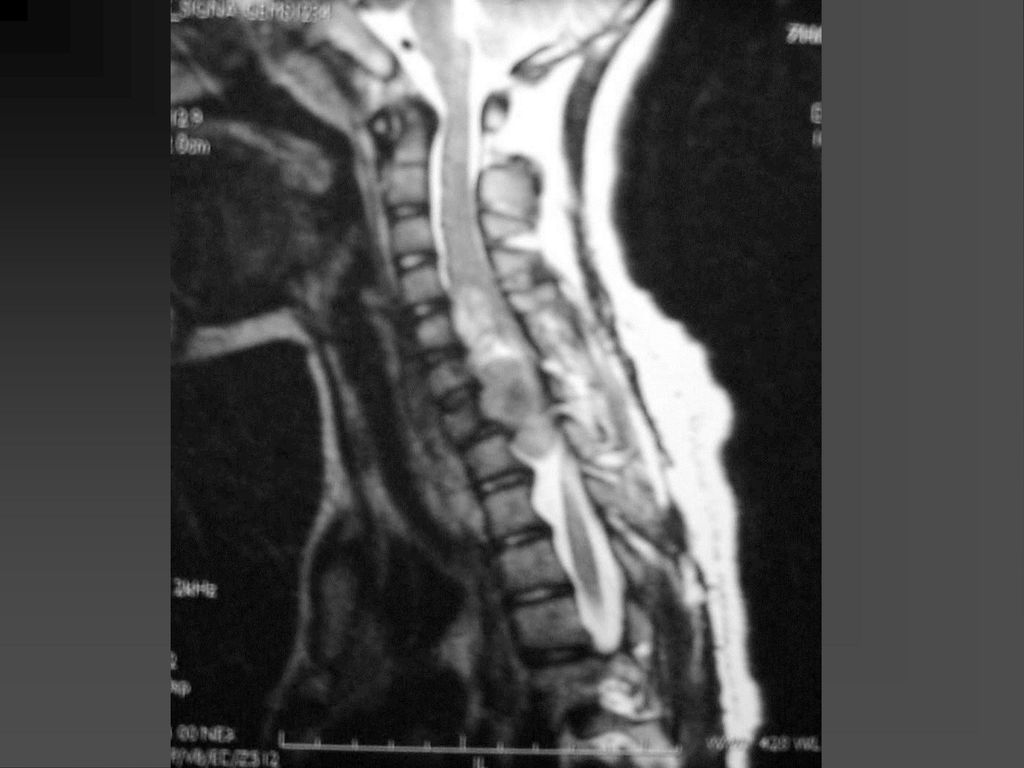
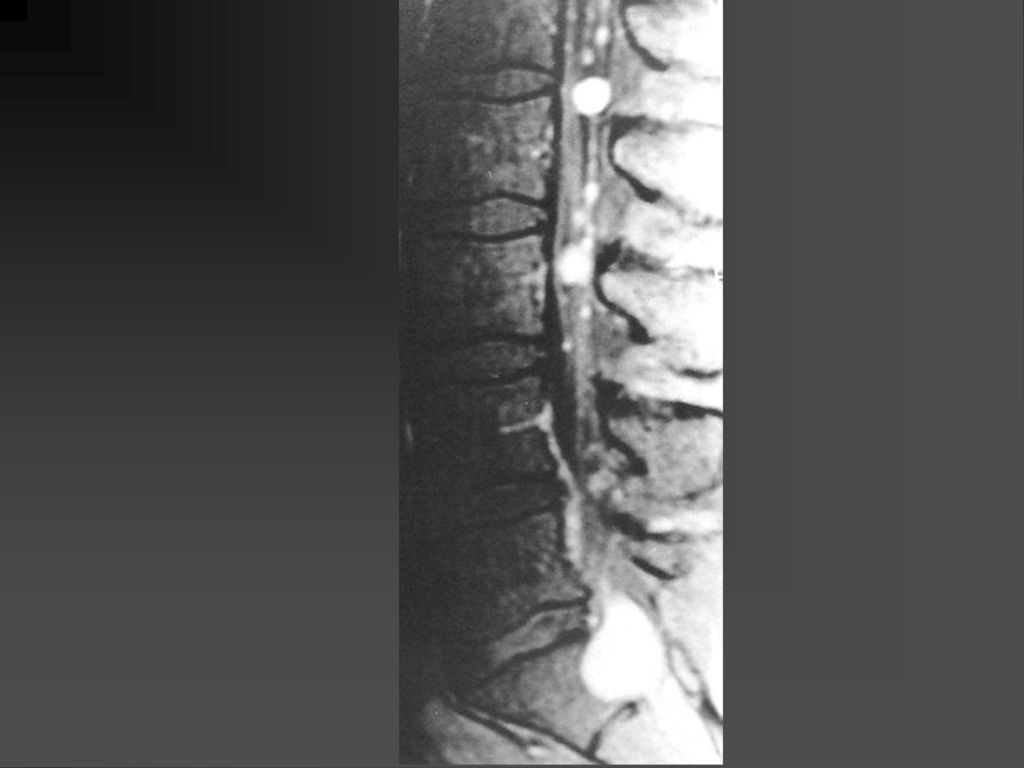
脊柱表现 脊柱侧弯,其发病率随年龄逐渐上升。 椎体后缘呈花边样改变,脊膜向两侧膨出,上述表现可能与中胚层发育不良有关。
偶尔可发生髓内星形细胞瘤,硬膜下神经纤维瘤。

42
神经纤维瘤病II型

43
诊断标准 凡符合以下两种标准之一即能作出诊断 (1)两侧性听神经瘤(无需活检,CT或MRI即能作出诊断)
(2)一级亲属患有此病加上单侧听神经瘤或加上以下两种肿瘤如其他颅神经雪旺氏细胞瘤、脑膜瘤或脊膜瘤、神经纤维瘤、胶质瘤等。
一级亲属患有此病加上单侧听神经瘤或加上以下两种肿瘤如其他颅神经雪旺氏细胞瘤、脑膜瘤或脊膜瘤、神经纤维瘤、胶质瘤等。")
44
临床表现 发生率占神经纤维瘤病的10% 儿童和成人均可发病,神经系统症状的出现常见青春期后,通常在20岁后。 约44%出现听力损害
皮肤异常改变远较神经纤维瘤病I型少见 半数以上的儿童患白内障。 儿童皮肤神经纤维瘤加上青少年白内障是检出神经纤维瘤病II型的重要线索。

45
病因及病理学 神经纤维瘤病II型也属常染色体显性遗传性疾病,具体表现在染色体22的异常。

46
影像学表现 神经纤维瘤病II型的颅内肿瘤以前庭神经鞘瘤为常见,以双侧多见。 脑膜瘤次之,可以是单发或多发,常位于幕上矢状窦旁及大脑凸面。
其他颅神经瘤依次是动眼神经、面神经、三叉神经等。 听神经瘤平扫桥小脑角区等低密度肿块、增强扫描较小肿瘤可为明显均匀强化,但较大肿瘤往往可见不强化的坏死囊变区,肿瘤大都向内听道延伸,内听道扩大。

55
其他颅脑表现 表现一些非肿瘤性颅内钙化,如脉络膜丛钙化、小脑皮质偶尔大脑表面皮质钙化。

56
脊柱表现 较常见的为椎管内及椎旁神经鞘瘤 最常见的髓内肿瘤是室管膜瘤 髓外硬膜下脊膜瘤也不少见。

58
结节性硬化 结节性硬化是一种家族性遗传性神经皮肤综合征,70%有基因突变。 包括了多系统的损害如神经、皮肤、眼、肾脏、心、肺等器官的损害

59
病因和病理 它是一种常染色体显性遗传性疾病,但是约50%~70%患者有基因突变。
90%患者有室管膜下或皮层结节,这两种结节病理学上有所不同,前者主要由巨细胞、神经、胶质以及血管等成分组成,后者同时还有胶质增生和髓鞘形成不良。 皮质结节往往发生在巨脑回样皮层中,这种皮质失去了正常的结构。 室管膜下巨细胞性星形细胞瘤由室管膜下结节发展而来,组织学表现界于错构瘤和巨细胞性肿瘤之间,属良性脑内肿瘤,偶可恶变。

60
临床表现 结节性硬化发病率为1:10000~20000,无明显的种族与性别差异。
典型的临床三联征是癫痫、智力低下、面部皮肤皮脂腺瘤(80%婴幼儿患者仅表现为婴儿痉挛和肌阵挛,智力低下发生率为45%~82%,90%患者有皮肤损害表现)。 15%病人可有视网膜错构瘤(即星形细胞增生) 40%~80%有肾脏的错构瘤。若并发室管膜下巨细胞性星形细胞瘤引起脑积水,则患者表现颅高压症状。
。 15%病人可有视网膜错构瘤(即星形细胞增生) 40%~80%有肾脏的错构瘤。若并发室管膜下巨细胞性星形细胞瘤引起脑积水,则患者表现颅高压症状。")
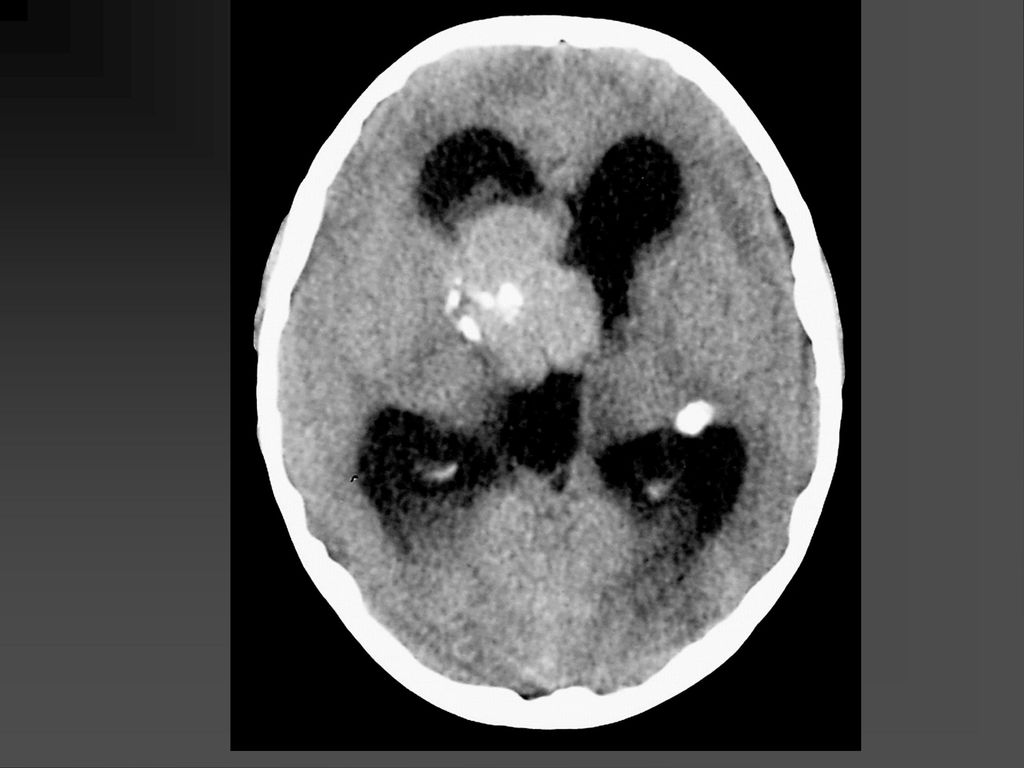
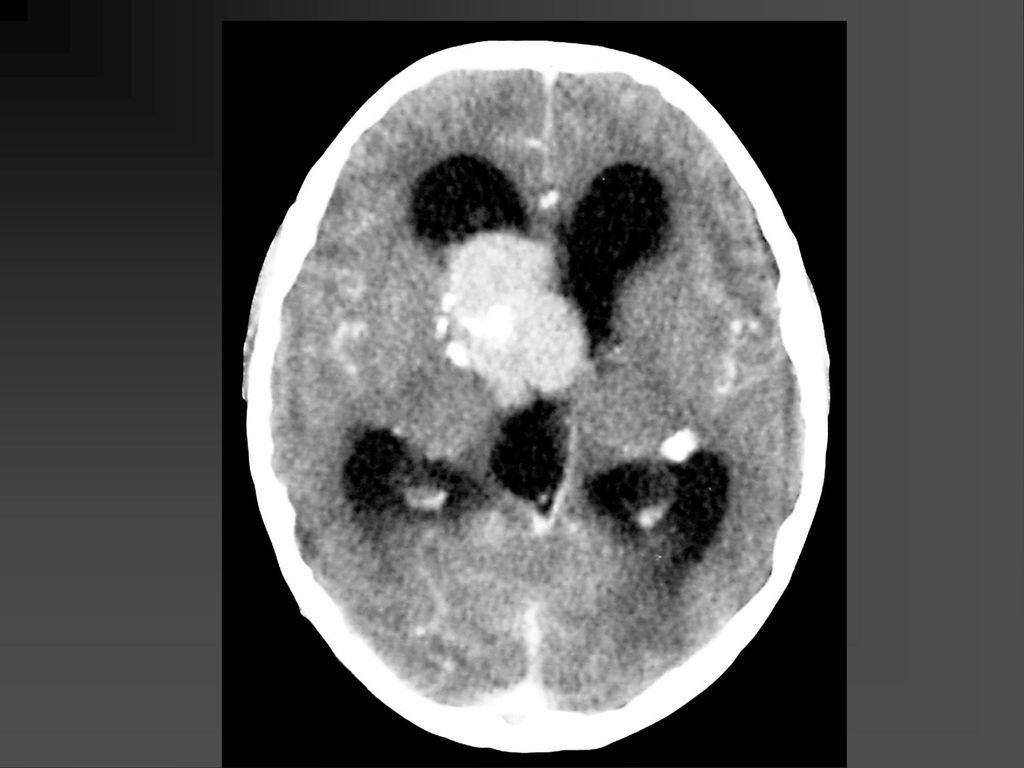
61
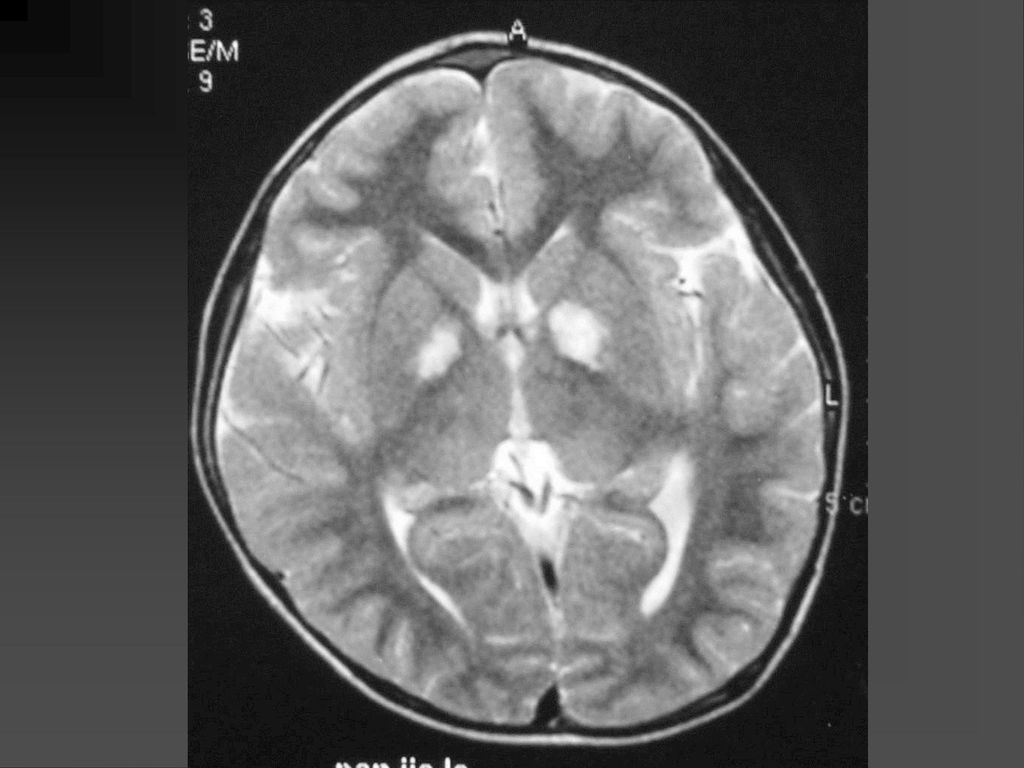
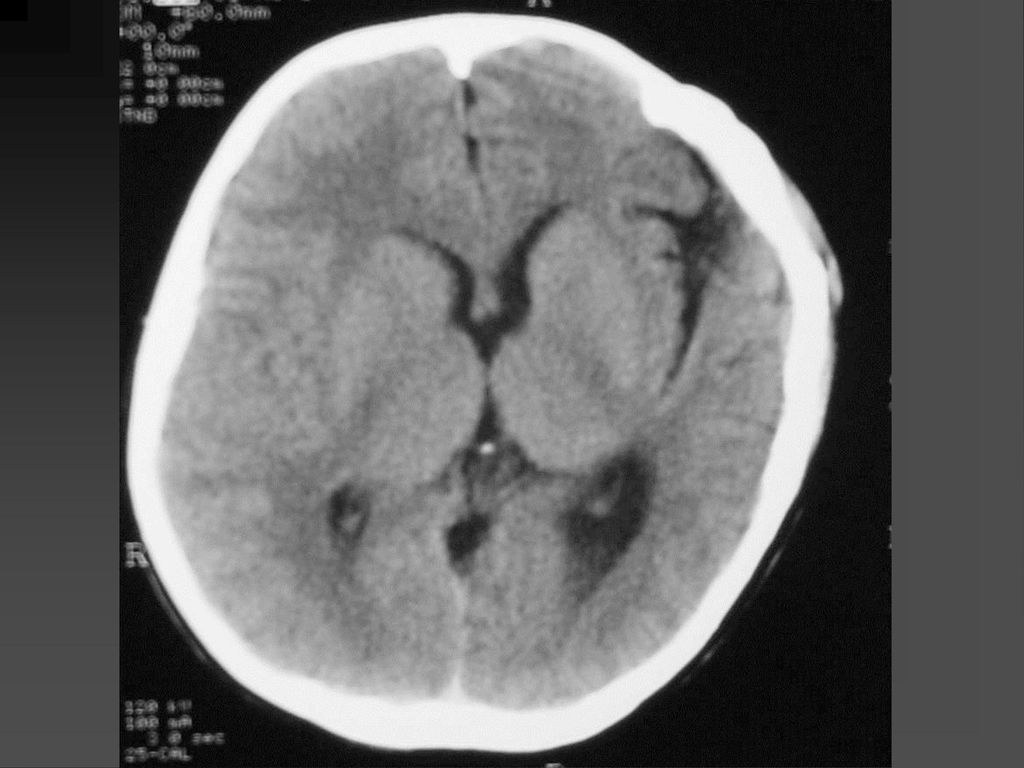
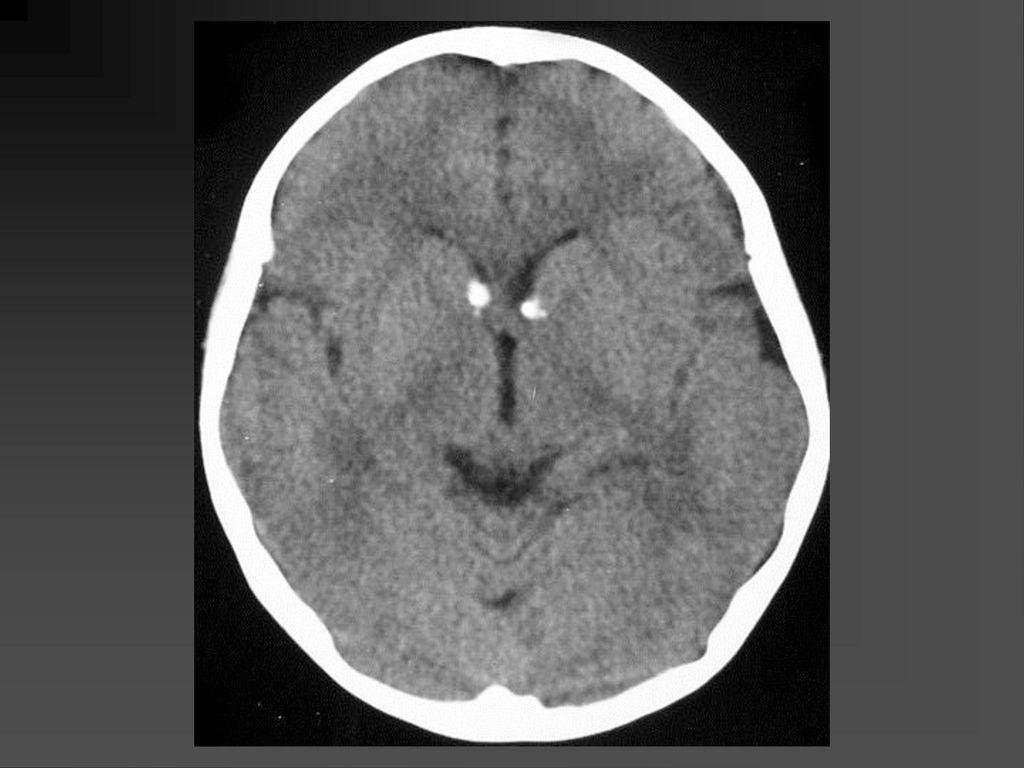
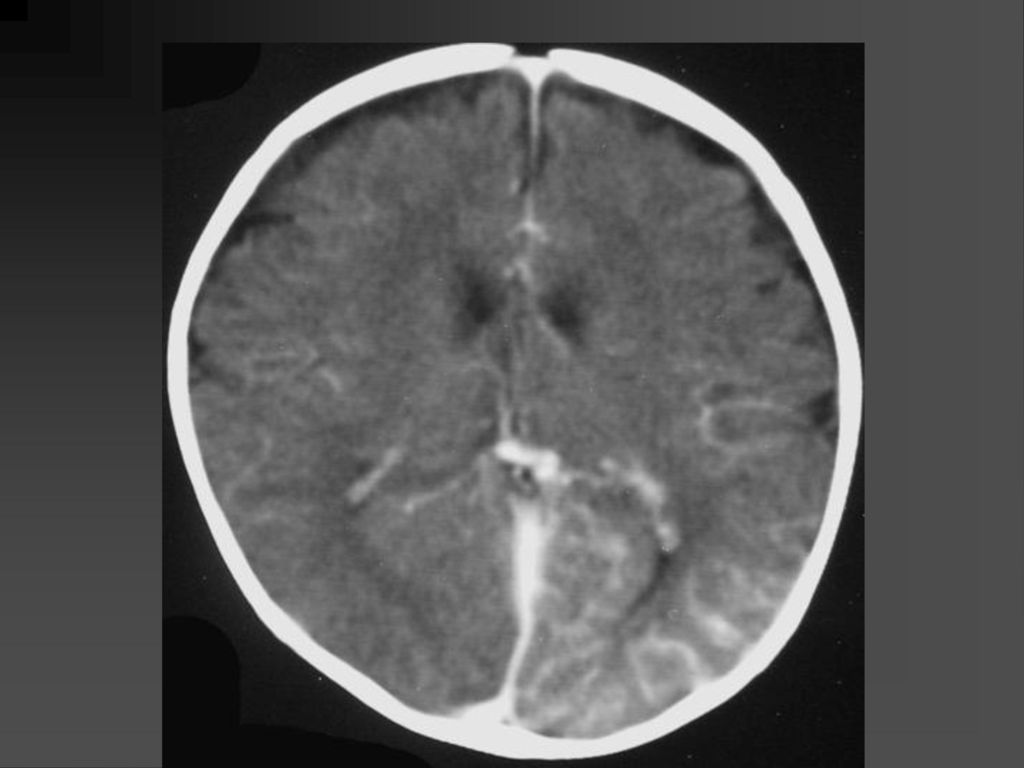
CT表现 脑特征性改变是室管膜下、皮层、皮层下结节 室管膜下结节见于侧脑室的外侧壁,Monro’s孔 附近最常见。

62
CT表现 室间孔处结节可发展成室管膜下巨细胞性星形细胞瘤,压迫室间孔引起梗阻性脑积水
若室管膜下结节直径超过12mm,该结节可能已经变成巨细胞性肿瘤,或随访检查结节逐渐增大,此时结节变成巨细胞性肿瘤可能性更大。 CT平扫肿瘤以等密度为主,常伴粗大钙化,增强扫描肿瘤明显强化。若肿瘤明显侵犯脑实质,则提示肿瘤可能为恶性。

68
MRI表现 室管膜下结节T1WI等信号或高信号、T2WI等信号,显示结节强化优于CT。
对钙化不敏感

71
脑颜面血管瘤综合征 (Sturge-Weber ’s syn.)
")
72
病因和病理 主要由多发小静脉缠绕在一起形成的软脑膜静脉瘤所组成,位于脑表面,而且有形成纤维化的趋势。
由于软脑膜表浅静脉发育不良或血栓形成,可出现脑室周围脑白质内粗大的深髓静脉和脑室内脉络膜丛增大。 皮质钙化是另一个重要的病理表现,主要出现在静脉瘤下的脑组织内,常见部位是颞顶枕,尤以枕叶为著。 整个大脑半球或两侧性受累较少见。

73
临床表现 绝大多数患者有面部葡萄酒色痣,以单侧多见 90%以上患者一岁内就有癫痫,患者可出现偏瘫 大多数病人智力落后
30%病人有眼球巩膜和脉络膜血管瘤,临床表现为眼痛、眶后痛 患侧脑萎缩严重者可见同侧颅骨发育小于对侧。 Sturge-Weber综合征一般没有内脏受累,有些病人伴有内脏血管瘤。

74
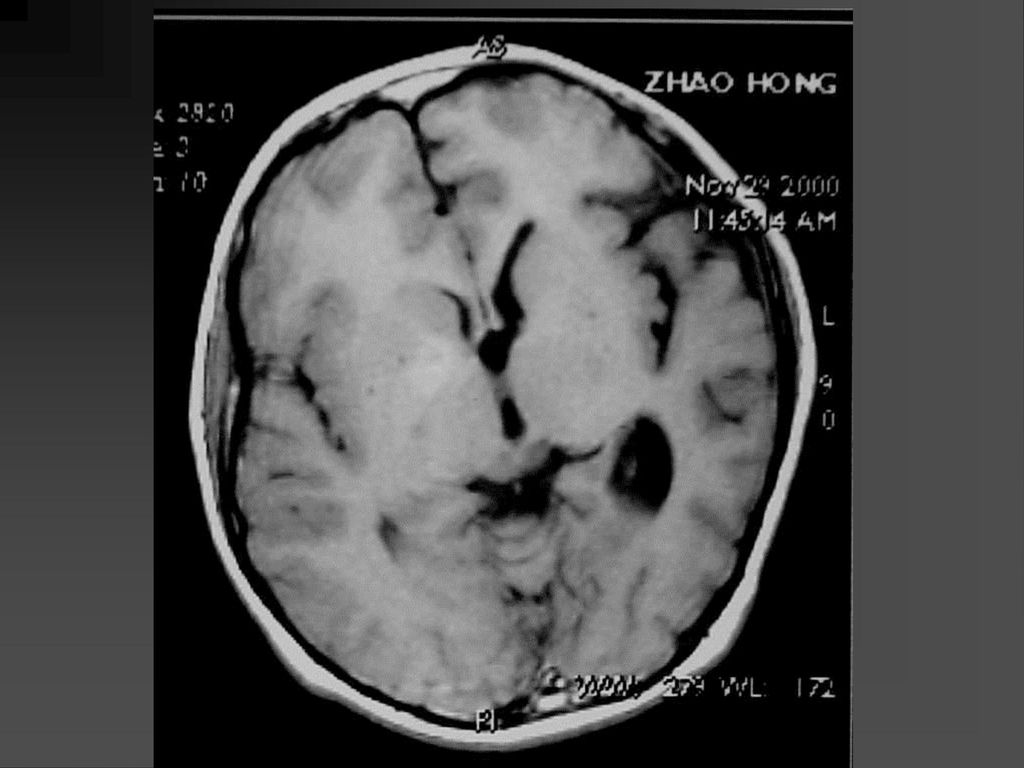
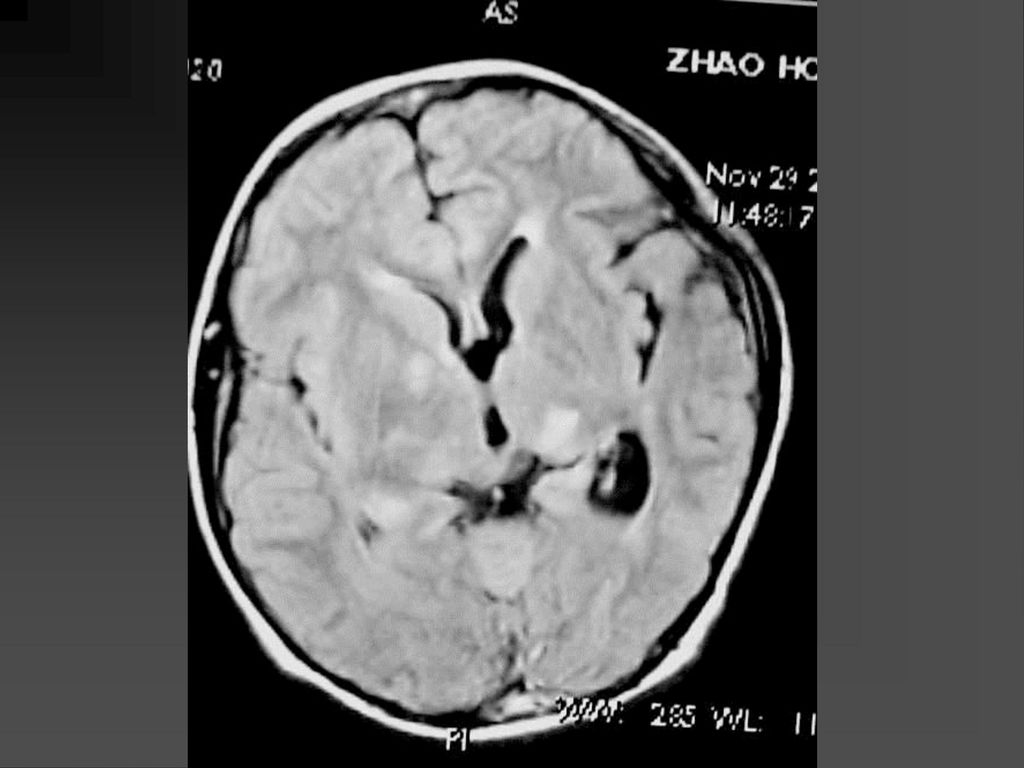
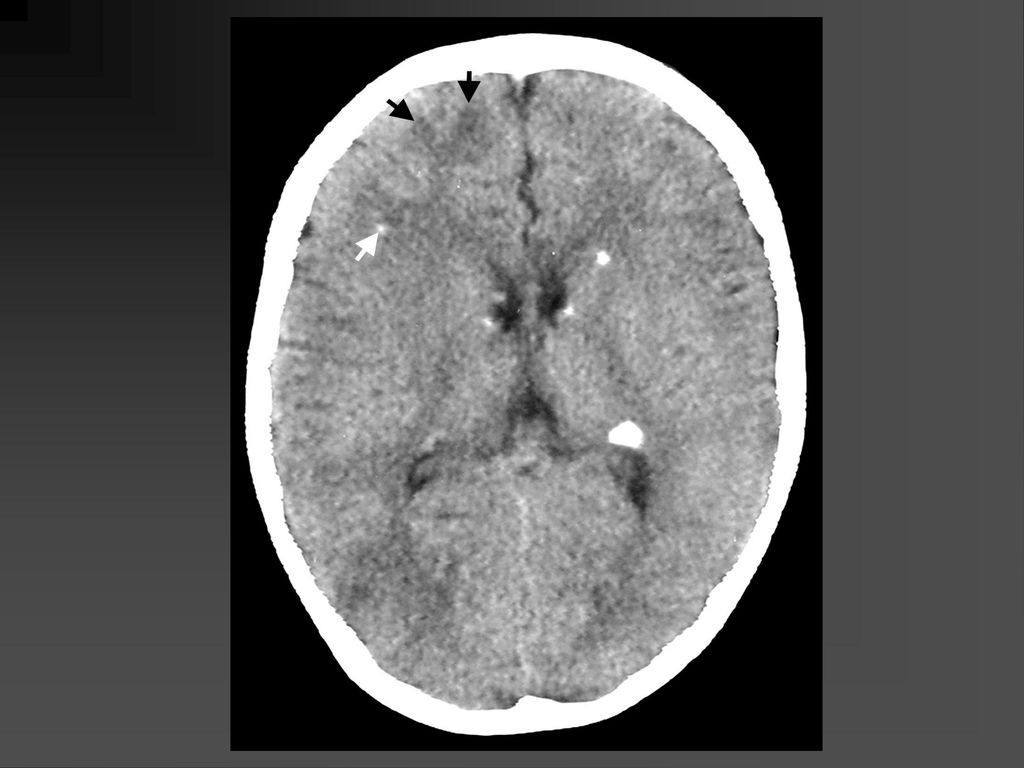
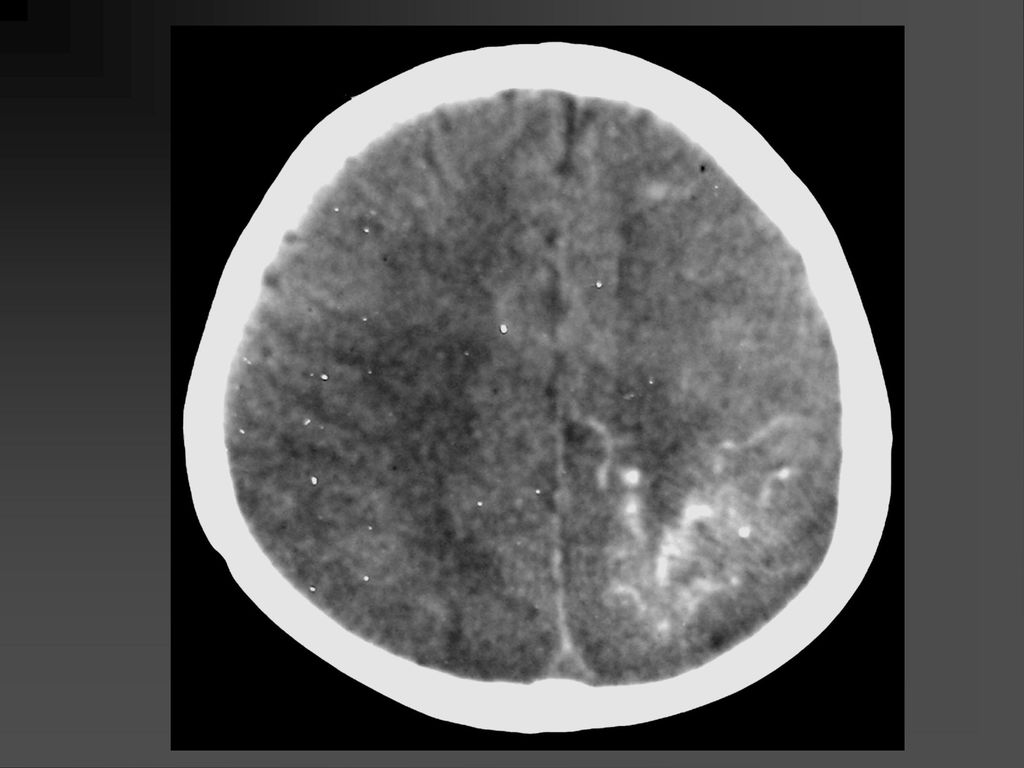
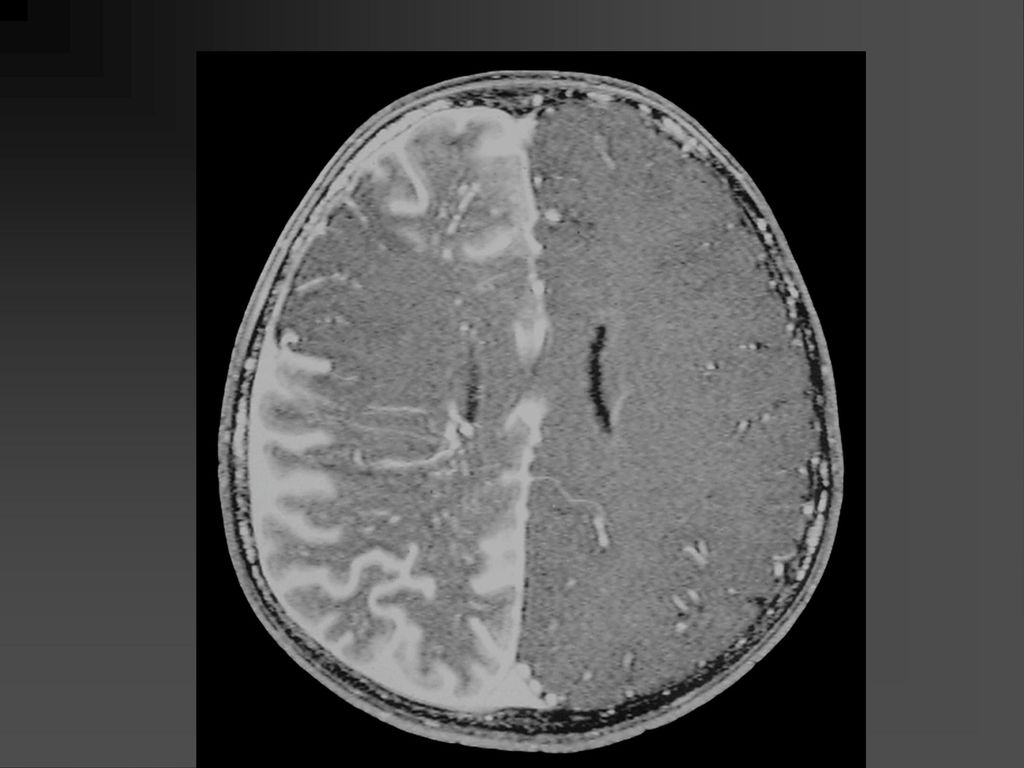
影像学表现 CT平扫可见颞顶枕部脑表面弧线样或脑回样钙化,一侧广泛性或两侧性钙化较少。
增强扫描在婴儿早期钙化尚未出现时表现为明显的脑回样强化及脉络膜丛异常强化,但随着皮质钙化的出现,增强表现常被掩盖。 MRI明显优于CT,它的强化不受钙化影响。 患侧的脉络丛常大于对侧。 患者常伴同侧的局限或广泛性脑萎缩表现,患侧 颅腔缩小。

80
血管母细胞瘤病 又称von Hippel-Lindau 综合征
诊断标准:(1).两个以上血管母细胞瘤(一个位于中枢神经系统、一个位于视网膜,或两个均位于中枢神经系统)。(2)一个中枢神经系统血管母细胞瘤加上相应内脏受累。(3)阳性家族史加一个血管母细胞瘤或内脏受累。
.两个以上血管母细胞瘤(一个位于中枢神经系统、一个位于视网膜,或两个均位于中枢神经系统)。(2)一个中枢神经系统血管母细胞瘤加上相应内脏受累。(3)阳性家族史加一个血管母细胞瘤或内脏受累。")
81
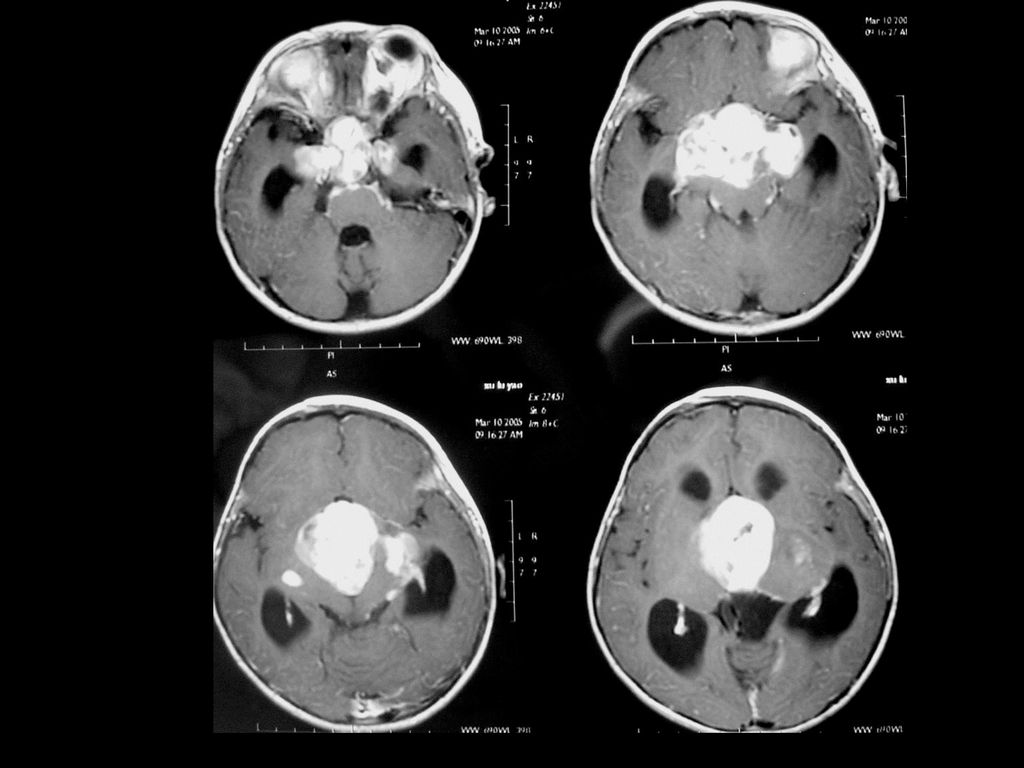
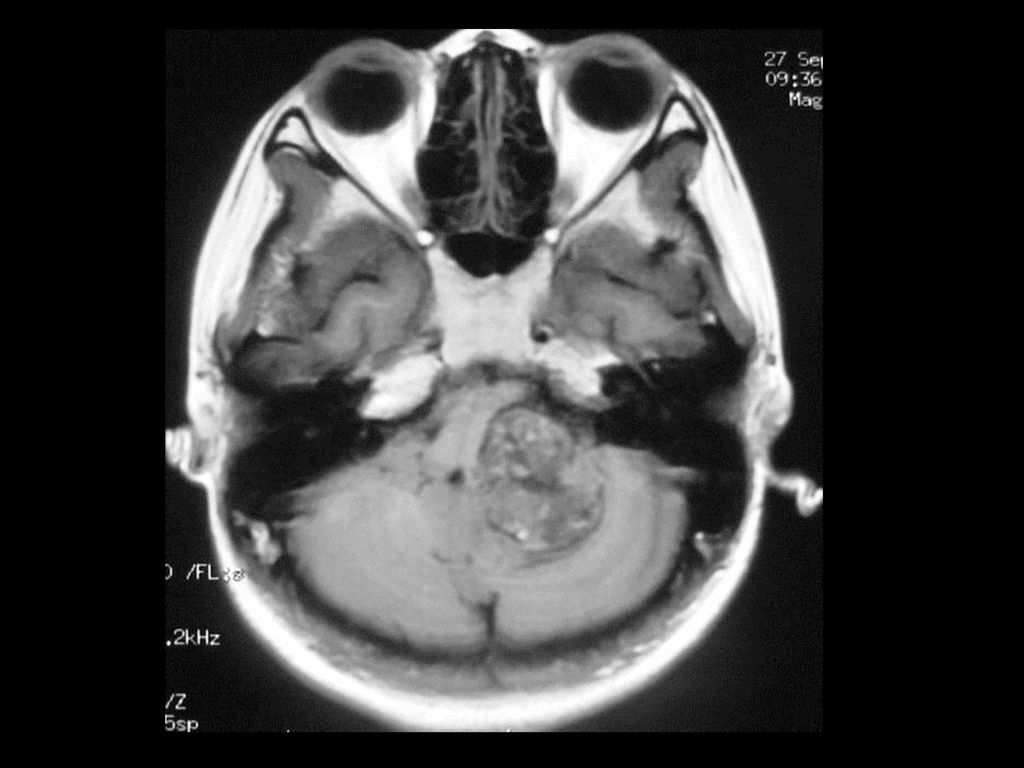
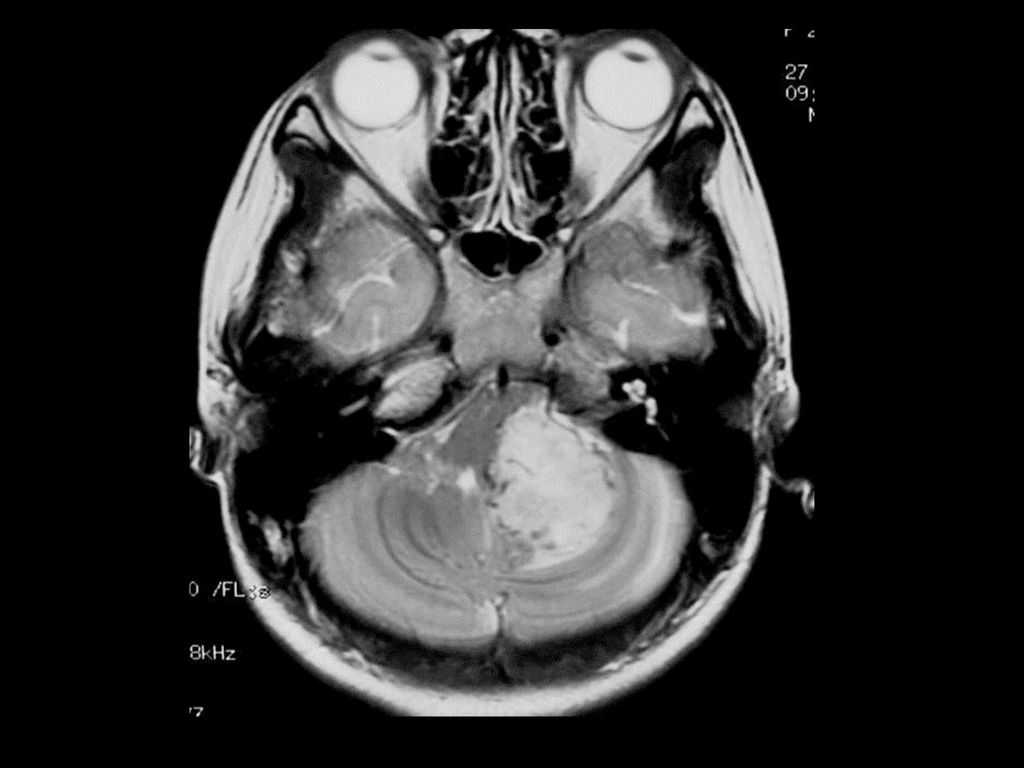
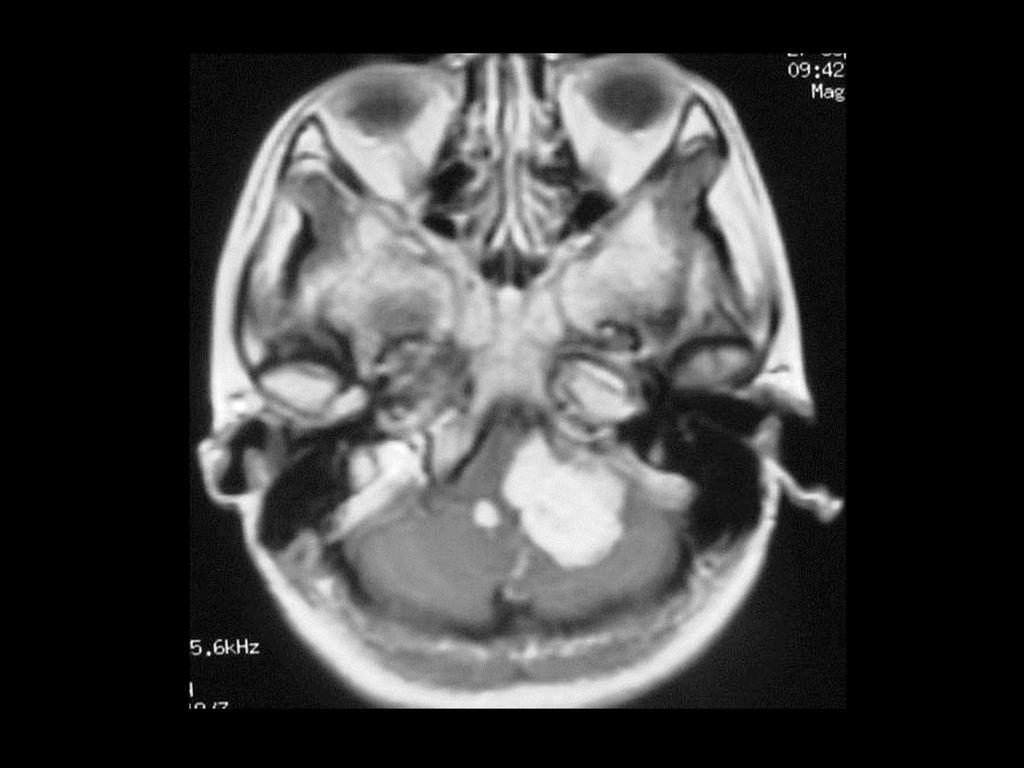
临床及影像学表现 常位于小脑半球,其次是延脑、颈髓。 小的血管母细胞瘤CT可能漏诊 典型表现为大囊小结节型,少数为实质型
增强扫描结节明显强化,实质部分与软脑膜相贴。 MRI可见肿瘤内明显扭曲的血管。