Download presentation
1
第三章 化学动力学基础 §3.1 化学反应速率的概念 §3.2 浓度对反应速率的影响 —速率方程 §3.3 温度对反应速率的影响
§3.1 化学反应速率的概念 §3.2 浓度对反应速率的影响 —速率方程 §3.3 温度对反应速率的影响 —Arrhenius方程 §3.4 反应速率理论和反应机理简介 §3.5 催化剂与催化作用

2
§3.1 化学反应速率的概念 平均速率和瞬时速率 定容反应速率

3
3.1.1 平均速率和瞬时速率 1. 平均速率 某一有限时间间隔内浓度的变化量。 2NO2 (CCl4) + O2(g)
平均速率和瞬时速率 1. 平均速率 某一有限时间间隔内浓度的变化量。 2NO2 (CCl4) + O2(g) 例: N2O5(CCl4)
 + O2(g) 例: N2O5(CCl4)")
4
40℃,5.00mLCCl4中N2O5的分解速率 N2O5(CCl4) 2NO2(CCl4)+ 1/2O2(g)
 2NO2(CCl4)+ 1/2O2(g)")
5
t1= 0 s c1(N2O5) = mol·L-1 t2=300 s c2(N2O5) = mol·L-1
 = mol·L-1 t2=300 s c2(N2O5) = mol·L-1")
6
时间间隔Δt趋于无限小时的平均速率的极限。
2. 瞬时速率 时间间隔Δt趋于无限小时的平均速率的极限。 c(N2O5)/(molL-1) t/s 经过A点切线斜率的负数为2700s时刻的瞬时速率。
/(molL-1) t/s. 经过A点切线斜率的负数为2700s时刻的瞬时速率。")
7
t1 = 0 s c1(N2O5)= mol·L-1 t2 = 5580 s c2(N2O5)= 0 mol·L-1 A点切线的斜率=
= mol·L-1 t2 = 5580 s c2(N2O5)= 0 mol·L-1 A点切线的斜率=")
8
定容反应速率 2NO2 (CCl4) + O2(g) 例: N2O5(CCl4)
 + O2(g) 例: N2O5(CCl4)")
9
对于一般的化学反应: r——定容条件下的反应速率(mol·L-1·s-1)
")
10
溶液中的化学反应: aA(aq) + bB(aq) yY(aq) + zZ(aq) 对于定容的气相反应:
 + bB(aq) yY(aq) + zZ(aq) 对于定容的气相反应:")
11
§3.2 浓度对反应速率的影响 ——速率方程 3.2.1 化学反应速率方程 3.2.2 由实验确定反应速率方程的 简单方法—初始速率法
§3.2 浓度对反应速率的影响 ——速率方程 化学反应速率方程 由实验确定反应速率方程的 简单方法—初始速率法 浓度与时间的定量关系

12
3.2.1 化学反应速率方程 40℃,CCl4中N2O5分解反应的r:c( N2O5 )
化学反应速率方程 40℃,CCl4中N2O5分解反应的r:c( N2O5 ) N2O5的分解速率与N2O5浓度的比值是恒定的,即反应速率r与c(N2O5)成正比。 可见:
 N2O5的分解速率与N2O5浓度的比值是恒定的,即反应速率r与c(N2O5)成正比。 可见:")
13
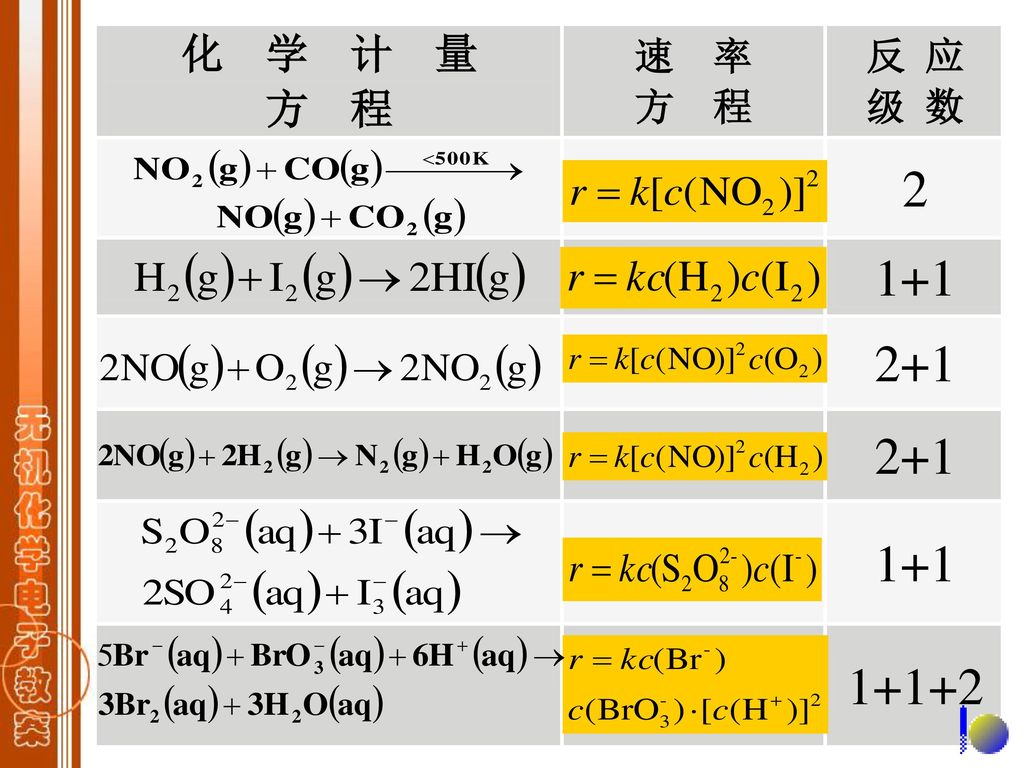
对于一般的化学反应: α,β—反应级数:若α=1,A为一级反应; β=2,B为二级反应,则α+β=3,总 反应级数为3。α,β必须通过实验确定其值。通常α≠a,β≠b。 k —反应速率系数:零级反应 mol·L-1 ·s-1; 一级反应 s-1;二级反应 (mol·L -1)-1 ·s-1。k 不随浓度而变,但受温度的影响,通常温度升高, k 增大。
-1 ·s-1。k 不随浓度而变,但受温度的影响,通常温度升高, k 增大。")
15
3.2.2 由实验确定反应速率方程的 简单方法—初始速率法
由实验确定反应速率方程的 简单方法—初始速率法 例如: 反应的有关实验数据如下:

16
该反应的速率方程: 对NO而言是二级反应,对H2而言是一级反应。 试问如何求出反应速率系数?

17
浓度与时间的定量关系 亦可写为:

18
lnc-t 关系应为直线

19
半衰期: 当反应物A的转化率为50%时所需的反应时间称为半衰期,用 表示。 对于一级反应,其半衰期为: ln ln 则 ln

20
零级、一级、二级反应的速率方程总结: * t1/2 *仅适用于只有一种反应物的二级反应。

21
§3.3 温度对反应速率的影响 —Arrhenius方程

22
3.3.1 Arrhenius方程 反应速率方程 影响反应速率的因素有: k和cB
k与温度有关,T增大,一般k也增大, 但k~T不是线性关系。

24
Ea—实验活化能,单位为kJ·mol-1。
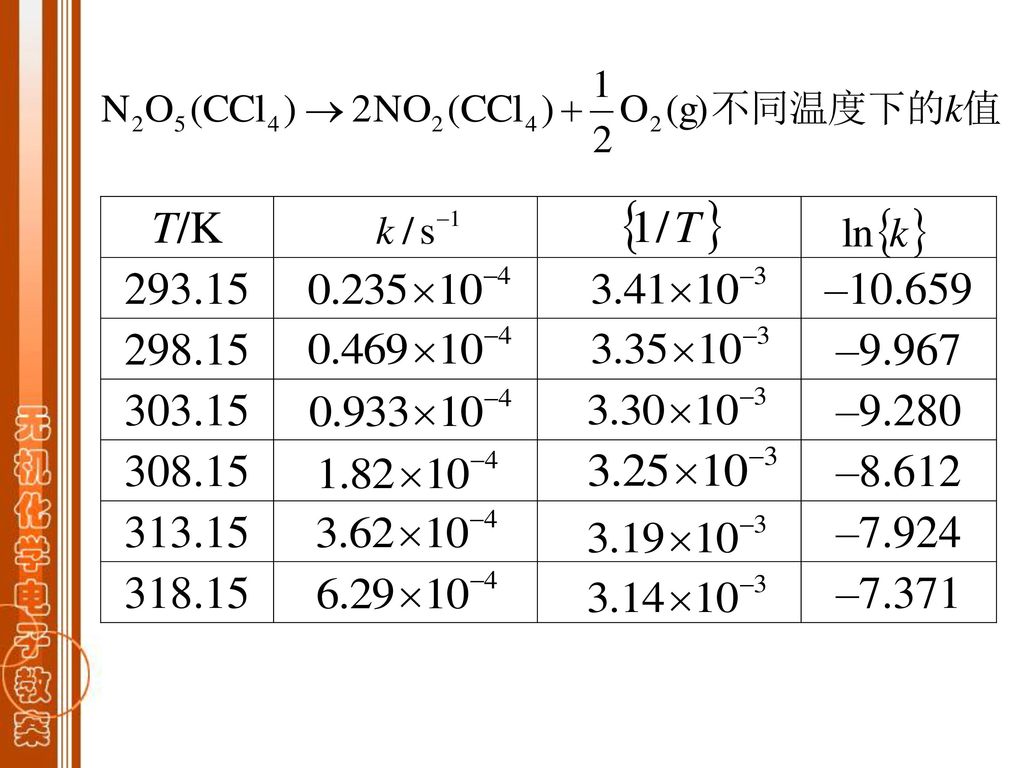
k-T 关系图: lnk-1/T 图 k-T 图 Arrhenius方程: (指数形式) k0—指前参量 Ea—实验活化能,单位为kJ·mol-1。
 k0—指前参量. Ea—实验活化能,单位为kJ·mol-1。")
25
显然ln{k}—{1/T}为直线关系, 直线的斜率为 , 直线的截距为ln{k0} 。 由Arrhenius方程可定义Ea:

26
3.3.2 Arrhenius方程的应用 1.已知T1—k1, T2—k2,求Ea 两式相减,整理得到:
通常活化能的数值在40 ~400 kJ·mol-1 之间,多数为60~250 kJ·mol-1 。

27
2.由Ea计算反应速率系数 例题: N2O5(g)2NO2 (g) +1/2O2(g) 已知:T1=298.15K, k1=0.469×10-4s-1 T2=318.15K, k2=6.29×10-4s-1 求:Ea及338.15K时的k3。

28
对Arrhenius方程的进一步分析 1. 在 ,Ea处于方程的指数项中,对k有显著影响,在室温下, Ea每增加4kJmol-1,k值降低约80%; 2. 温度升高,k增大,一般反应温度每升高10℃,k将增大2~10倍;

29
3. 根据 对同一反应,升高一定温度,在高温区值增加较少, 因此对于原本反应温度不高的反应 ,可采用升温的方法提高反应速率;
4. 对不同反应,升高相同温度, Ea大的反应 k 增大的倍数多,因此升高温度对反应慢的反应有明显的加速作用。

30
§3.4 反应速率理论和反应机理简介 3.4.1 碰撞理论 3.4.2 活化络合物理论 3.4.3 活化能与反应速率
§3.4 反应速率理论和反应机理简介 碰撞理论 活化络合物理论 活化能与反应速率 反应机理与元反应

31
3.4.1 碰撞理论 以气体分子运动论为基础,主要用于气相双分子反应。 例如:反应 发生反应的两个基本前提: 发生碰撞的分子应有足够高的能量
碰撞理论 以气体分子运动论为基础,主要用于气相双分子反应。 例如:反应 发生反应的两个基本前提: 发生碰撞的分子应有足够高的能量 碰撞的几何方位要适当

32
能够发生反应的碰撞为有效碰撞。 能够发生有效碰撞的分子为活化分子。

33
气体分子的能量分布和活化能

34
活化络合物理论(过渡态理论) 以量子力学对反应过程中的能量变化的研究为依据,认为从反应物到生成物之间形成了势能较高的活化络合物,活化络合物所处的状态叫过渡态。 例如反应: 其活化络合物为 ,具有较高的势能Eac 。它很不稳定,很快分解为产物分子NO2和O2。 N O O O O

36
化学反应过程中能量变化曲线

37
E(Ⅰ)-反应物(始态)势能 E(Ⅱ)-生成物(终态)势能 正反应的活化能 Ea(正) =Eac - E(Ⅰ) 逆反应的活化能 Ea(逆) =Eac - E(Ⅱ) ΔrHm= E(Ⅱ) - E(Ⅰ)= [Eac - Ea(逆)] -[Eac - Ea(正)] ΔrHm= Ea(正) - Ea(逆) Ea(正) <Ea(逆), ΔrHm <0 ,为放热反应; Ea(正) >Ea(逆), ΔrHm >0 ,为吸热反应。
![E(Ⅰ)-反应物(始态)势能 E(Ⅱ)-生成物(终态)势能. 正反应的活化能 Ea(正) =Eac - E(Ⅰ) 逆反应的活化能 Ea(逆) =Eac - E(Ⅱ) ΔrHm= E(Ⅱ) - E(Ⅰ)= [Eac - Ea(逆)] -[Eac - Ea(正)]](http://slidesplayer.com/slide/11444985/61/images/37/E%28%E2%85%A0%29%EF%BC%8D%E5%8F%8D%E5%BA%94%E7%89%A9%28%E5%A7%8B%E6%80%81%29%E5%8A%BF%E8%83%BD+E%28%E2%85%A1%29%EF%BC%8D%E7%94%9F%E6%88%90%E7%89%A9%28%E7%BB%88%E6%80%81%29%E5%8A%BF%E8%83%BD.+%E6%AD%A3%E5%8F%8D%E5%BA%94%E7%9A%84%E6%B4%BB%E5%8C%96%E8%83%BD+Ea%28%E6%AD%A3%29+%3DEac+-+E%28%E2%85%A0%29+%E9%80%86%E5%8F%8D%E5%BA%94%E7%9A%84%E6%B4%BB%E5%8C%96%E8%83%BD+Ea%28%E9%80%86%29+%3DEac+-+E%28%E2%85%A1%29+%CE%94rHm%3D+E%28%E2%85%A1%29+-+E%28%E2%85%A0%29%3D+%5BEac+-+Ea%28%E9%80%86%29%5D+-%5BEac+-+Ea%28%E6%AD%A3%29%5D.jpg "ΔrHm= Ea(正) - Ea(逆) Ea(正) <Ea(逆), ΔrHm <0 ,为放热反应; Ea(正) >Ea(逆), ΔrHm >0 ,为吸热反应。")
38
3.4.3 活化能与反应速率 Arrhenius活化能:由普通分子转化为活化分子所需要的能量。 Tolman活化能:
活化能与反应速率 Arrhenius活化能:由普通分子转化为活化分子所需要的能量。 Tolman活化能: 对于气相双分子简单反应,碰撞理论已推算出:Ea=Ec+1/2RT≈Ec 。 常把Ea看作在一定温度范围内不受温度的影响。

39
浓度影响:当温度一定,某反应的活化能也一定时, 浓度增大, 分子总数增加,活化分子数随之增多,反应速率增大。
温度影响:当浓度一定,温度升高,活化分子分数增多, 反应速率增大。

40
3.4.4 反应机理与元反应 反应机理:化学反应过程中经历的真实反应步骤的集合。
反应机理与元反应 反应机理:化学反应过程中经历的真实反应步骤的集合。 元反应:由反应物一步生成生成物的反应,没有可用宏观实验方法检测到的中间产物。 意义:通过实验一旦证实某一有确定 反应物和生成物的反应为元反应,就可以根 据化学反应计量方程式直接写出其速率方 程式。 例如: 为元反应 则

41
复合反应:由两个或两个以上的反应组合而成的总反应。在复合反应中,可用实验检测到中间产物的存在,但它被后面的一步或几步反应消耗掉,因而不出现在总反应方程式中。
如: 为由下列两步组成的复合反应 (慢) (快) 中间产物NO3可被光谱检测到,但是没有从混合物中分离出来。 控制步骤的速率方程式:
 (快) 中间产物NO3可被光谱检测到,但是没有从混合物中分离出来。 控制步骤的速率方程式:")
42
反应机理的研究是一个十分复杂而艰难的任务。 意义:若清楚反应是如何进行的,则可以有效控制反应的快慢,以获得期望产物。
一般的过程是:采用分子光谱等研究手段检测反应过程中的中间产物,据此推断反应历程,再以实验获得的速率方程验证。 一个合理的反应机理应满足: 全部元反应的加和应为化学计量反应方程式 由反应机理得出的速率方程应与实验所得一致

43
O N NO ① 例题:一氧化氮被还原为氮气和水: 根据光谱学研究提出的反应机理是: k1 (快, 平衡) k-1 k2 (慢) k1
(快, 平衡) 2 O N NO ① k1 k-1 (慢) k2 (快) k1 依据这一反应机理推断其速率方程式,并确定相关物种的反应级数。
 2. O. N. NO. ①. k1. k-1. (慢) k2. (快) k1. 依据这一反应机理推断其速率方程式,并确定相关物种的反应级数。")
44
解:按照速率控制步骤(最慢的一步) 是中间产物,根据第一步的快速平衡, 则 代入 该反应对NO是二级反应,对H2是一级反应。
 是中间产物,根据第一步的快速平衡, 则 代入 该反应对NO是二级反应,对H2是一级反应。")
45
§3.5 催化剂与催化作用 催化剂和催化作用的基本特征 均相催化与多相催化 酶催化

46
催化剂和催化作用的基本特征 催化剂:存在少量就能加快反应而本身最后并无损耗的物质。

47
催化作用的特点 : ①只能对热力学上可能发生的反应起作用。 ②通过改变反应途径以缩短达到平衡的时间。 ③催化剂有选择性,选择不同的催化剂会有 利于不同种产物的生成。 ④只有在特定的条件下催化剂才能表现活性。

48
3.5.2 均相催化与多相催化 1.均相催化: 催化剂与反应物种在同一相中的催化反应。 没有催化剂存在时,过氧化氢的分解反应为:
均相催化与多相催化 1.均相催化: 催化剂与反应物种在同一相中的催化反应。 没有催化剂存在时,过氧化氢的分解反应为: 加入催化剂Br2,可以加快H2O2分解,分解反应的机理是: 第一步 第二步 总反应:

49
反应历程 催化剂对反应活化能的影响

50
实验结果表明,催化剂参与的分解反应,改变了反应机理,降低了反应活化能,增大了活化分子分数,反应速率显著增大。
有催化 无催化 活化能降低使活化分子分数增加 实验结果表明,催化剂参与的分解反应,改变了反应机理,降低了反应活化能,增大了活化分子分数,反应速率显著增大。

51
2.多相催化: 催化剂与反应物种不属于同一物相的催化反应。 汽车尾气(NO和CO)的催化转化: 反应在固相催化剂表面的活性中心上进行,催化剂分散在陶瓷载体上,其表面积很大,活性中心足够多,尾气可与催化剂充分接触。
的催化转化: 反应在固相催化剂表面的活性中心上进行,催化剂分散在陶瓷载体上,其表面积很大,活性中心足够多,尾气可与催化剂充分接触。")
52
酶催化 酶催化:以酶为催化剂的反应。 特点:①高效 ②高选择性 ③条件温和












的减少量或生成物(浓度)的增大量表示>")
积分形式 (2)动力学特征 (3)应用举例>")
 常数项级数的概念 袁安锋 2016.7.>")







