Download presentation
1
高等有机化学 物理有机化学 有机合成反应 金属有机化学 高等有机化学与基础有机化学的差异: 物理有机化学与理论有机化学
有机合成反应与有机合成设计技巧 金属有机化学,有机金属参与的在机反应 物理有机化学 有机合成反应 金属有机化学

2
有机化学(专业)核心期刊/主流杂志 有机合成: 原子经济、环境、成本、不对称合成等.
Organic Letter (ACS) IF = 3.9 Journal of Organic Chemistry (ACS) IF = 3.6 Oranometallics (ACS) IF = 3.2 Advanced Synthesis and Catalysis IF = 3.2 Green Chem IF=2.82 Tetrahedron Letter IF = 2.4 Synthesis Letter IF = 2.7 Tetrahedron IF = 2.4 Tetrahedron-Asymmetry IF = 2.4 Europeal Journal of Organic Chemistry IF = 2.2 Synthese-Stuttgart IF = 2.2 有机合成: 原子经济、环境、成本、不对称合成等.
 IF = 3.9. Journal of Organic Chemistry (ACS) IF = 3.6. Oranometallics (ACS) IF = 3.2. Advanced Synthesis and Catalysis IF = 3.2. Green Chem. IF=2.82. Tetrahedron Letter IF = 2.4. Synthesis Letter IF = 2.7. Tetrahedron IF = 2.4. Tetrahedron-Asymmetry IF = 2.4. Europeal Journal of Organic Chemistry IF = 2.2. Synthese-Stuttgart IF = 2.2. 有机合成: 原子经济、环境、成本、不对称合成等.")
3
Top 化学杂志 Angew. Chem. Int. Edit. IF=8.43 J. Am. Chem. Soc. IF=6.52
Nature IF=30.98 Scicene IF=29.16 Chem Rev IF=21.04 Accounts Chem. Res IF=15.00 Chem. Soc. Rev IF=9.57 Angew. Chem. Int. Edit IF=8.43 J. Am. Chem. Soc IF=6.52 Nano Lett IF=6.14 Chem-A Eur. J IF=4.35 Chem. Comm IF=4.03

4
高等有机化学 高等有机化学又名物理有机和理论有机化学
研究对象:有机化合物的结构以及有机化合物在反应过程中结构的变化,研究有机分子的结构和反应条件对有机化合物的物理、化学性能的影响以及化学反应历程。 它的理论基础主要是量子化学和以此为依据的化学键理论和电子理论。

5
高等有机化学 通过对一般典型有机结构的性质及典 型反应历程的研究,使有机合成化学 家有可能运用这些理性认识来推测未
知有机物极其在反应中的内在联系, 从而有利于设计具有特殊性能的新化 合物,考虑合成中的最好原料和最理 想的合成路线等。

6
第一章 化学成键作用和分子结构 分子轨道法所用的量子理论,为分子提供了数学模型,这些数学模型也只能是真实分子的近似代表物,因为对所有有机分子的量子力学描述,都需要一定的近似才能求得它们的数学解。 成键理论定性和定量两个方面来描述化学成键作用

7
1. 1化学成键中的价键理论 1916年G.N.刘易斯(Lewis)提出化学成键作用:是两个原子间共享电子对的结果,他的这一提法是化学成键理论的一个质的飞跃4。 刘易斯的建议基本上还是直觉的,直到1927年海特勒(Haitler)和伦敦(London) 用量子力学对氢分子的处理才标志着价键理论的诞生5。
和伦敦(London) 用量子力学对氢分子的处理才标志着价键理论的诞生5。")
8
价键理论的特点 价键理论的一个重要的特点是:处于平衡核间距的两个原子间的结合能,大部分是由于两个核之间的电子交换(共振)而产生的。
而产生的。")
9
价键理论的局限 在氢分子中,如果认为电子1仅限于同核1相结合,而电子2仅限于同核2相结合,计算出来的结合能只是实测的键能的很小一部份。
如果取消这种限定,认为两个电子是不可区分的并同两个核同等地相互作用,则计算出来的结合能可以非常好地与实验测得的键能值相吻合。于是在氢分子H-H中用一条短线代表两个氢原子由于球形对称的1S轨道互相重叠而在两个核之间形成的一对电子占据的轨道。

10
价键理论的局限: 把成键电子描述成定域在两个原子之间,是一种定域的分子轨道理论。
在定量计算分子的性质时,价键法现已大部分被分子轨道法所代替。

11
1.1.1 杂化轨道 杂化轨道的原理是:原子在化合过程中为了形成的化学键强度更大,更有利于体系能量的降低,趋向于将原有的能量不同的原子轨道进一步线性组合成新的原子轨道,这只有在受到其它原子的“微扰”时才能发生,这种线性组合称为杂化,杂化后的原子轨道称为杂化轨道。

12
杂化轨道 用波谱法测量碳在基态时的电子构型为1s2,2s2,2p2,从价键理论的角度来看,它在形成化学键时,应该是二价的,但实际上它是四价的并且它的价键具有四面体取向。 如在甲烷中,碳原子上连有四个相同的配位体,它是一个正四面体,每个H-C-H键角都等于109.5°。

13
杂化轨道 为了解决碳的价键的四面体取向问题,L . 鲍林(Linus Pauling)发展了定向价和轨道杂化两个概念。
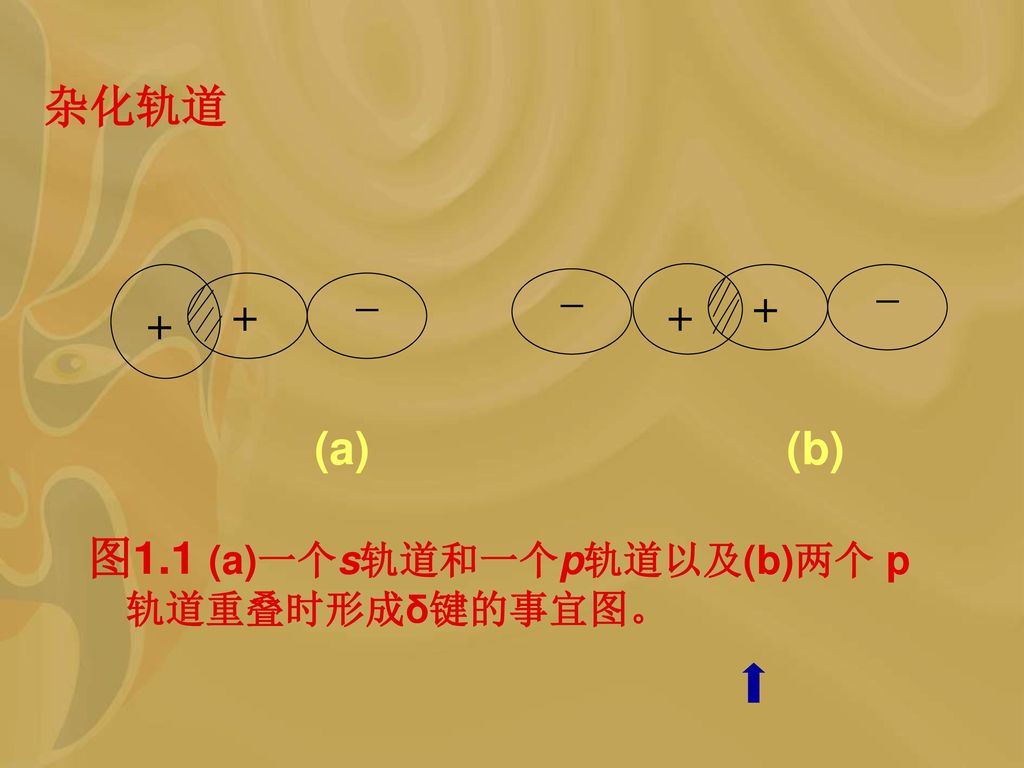
鲍林认为:由于共价键要求轨道相互重叠,所以较强的键是来自于较好的重叠。 在图1.1中绘示出一个原子的p轨道同另一个原子的s轨道或p轨道相重叠而在两个原子间形成共价键的情况。 结果是:电子的分布围绕核轴呈圆柱形对称,这种键定义为δ键。

14
杂化轨道 (a) (b) 图1.1 (a)一个s轨道和一个p轨道以及(b)两个 p轨道重叠时形成δ键的事宜图。 _ _ _ + + + +
15
杂化轨道 鲍林建议用四个等当的标记为sp3的杂化轨道来代替四个价轨道(2s,2px,2py,2pz)。这些轨道的近似形状于图1.2中。 这样碳就可以形成四个键而不是两个键。 sp3轨道的几率分布具有高度的方向性(最大几率区集中在核的一侧)为极其有效的重叠创造了条件。
。这些轨道的近似形状于图1.2中。 这样碳就可以形成四个键而不是两个键。 sp3轨道的几率分布具有高度的方向性(最大几率区集中在核的一侧)为极其有效的重叠创造了条件。")
16
杂化轨道 图1.2 轨道角向分布断面图

17
杂化轨道 四个等当sp3杂化轨道上各有一个电子的碳原子比光谱基态的碳原子具有较高的能量,但是使两个电子从2s轨道激发到sp3轨道形式上所需要的能量能够被形成四个键(而不是形成两个键)时释放出来的能量所补偿而有余,由于杂化轨道的方向性而使每个键都成为较强的键。 杂化作用的数学描述预示了碳具有四面体的几何形状。

18
杂化轨道 在乙烯和乙炔中,碳分别用sp2 和sp杂化轨道成键 碳原子采取不同的杂化形式和不同的配位数形成的分子具有不同的几何形状,它们之间的关系如表1.1所示。 甲烷和四氯化碳中的键角为109.5°,环己烷中的C-C-C键角为111.5°。在甲醛中的H-C-H键角为118°,而不是120°,不过苯是一个键角为120°的正六边形。

19
表1.1 结构随碳杂化形式变化的情况 配位体数目 杂化形式 几何形状 键 角 例 子 4 Sp3 四面体 109.5° 甲烷, 乙烷, 甲
配位体数目 杂化形式 几何形状 键 角 例 子 Sp 四面体 ° 甲烷, 乙烷, 甲 醇, 四氯化碳 Sp 三角形 ° 乙烯, 丙酮, 甲基正 离子, 碳酸根负离子 sp 线 形 ° 乙炔, 二氧化碳, 氰化氢

20
S 特征百分数: 与开链化合物相比,环丙烷、环丁烷和双环分子中的键角偏离正常值很多。
如在环丙烷中,三个碳原子处于等边三角形的顶角上,核间角为60°。 这种排布方式使这些环状化合物分子中引入了附加能量,这种能量称为角张力。 人们假定环丙烷中用于形成碳-碳键的轨道如果比它们正常的sp3轨道具有更多的p特征的话,则它们就能更有效地重叠,因为p特征的增大相应于键角的减少。这样一来,用来与氢成键的轨道就具有较多的s特征。

21
S 特征百分数: 为了定量地描述杂化的这种情况,人们给C-H键中的“s特征百分数”规定了一定数值。
在C-H键和C-C键的s特征百分数分别为33%和17%。 在环丙烷中最大轨道重叠区不在核轴上,并且C-C键为“弯键”(图1.5)。 核间角和轨道间角在大多数情况下是同义词,但在具有角张力的化合物中就不一样了,对于环丙烷来,核间角为60°,而轨道键角约为104°。
。 核间角和轨道间角在大多数情况下是同义词,但在具有角张力的化合物中就不一样了,对于环丙烷来,核间角为60°,而轨道键角约为104°。")
22
不等性杂化: 定义:由于孤对电子的存在而造成的不完全等同的杂化轨道过程,叫不等性杂化。
如水分子中氧原子上的两对孤对电子占据两个sp3杂化轨道,剩下的两个杂化轨道各含有一个未成对电子,所以能形成两个化学键,水分子的键角应为109.5°,和实验值105°有较大的偏差,这是由于原子上的孤对电子不参加成键作用,电子云较密集于该原子的周围,这样才有利于能量的降低。 因此,孤对电子所占据的杂化轨道具有较多的s成分,其它杂化轨道则含有比原来sp3杂化轨道较多的p轨道成分。

23
1.1.2 共振论 共振论代表价键理论的一种引伸,它用来处理能画出不止一个刘易斯结构的分子。 共振论的基本论点是:
如果一个分子或分子的一部分可以写出几种不同的刘易斯结构,而所有这些结构中原子核的相对位置是固定不变的,只是核间电子分布不同时,那么这个分子不宜用单独一种刘易斯结构来表示,它具有所有这些结构代表的性质。

24
共振论的基本论点 2. 有一些刘易斯结构比另一些更为合理。 最接近于真实分子的结构是具有下列特点 的结构:共价键的数目最多、异号电荷的
分离程度最低以及任何负电荷都处于电负 性最大的原子上(或任何正电荷都在电正 性最大的原子上)。
。")
25
当对一个共轭不饱和羰基化合物只能画出一个不带电荷的结构时,第二个带电荷的结构是重要的。
它把一个负电荷放在氧上,而给β碳原子留下一个正电荷。 可以预料到,第三个结构的贡献其重要性要小得多。这个结构中电荷的位置倒过来了,由于正电荷在氧上,所以这个结构是不稳定的。

26
当两种共振依据有矛盾时,就应作进一步的判断。
例如,对于在苯甲醚硝化反应的决定速度步骤中所形成的中间体来说,尽管刘易斯结构A有一个正电荷在氧上,但它是共振杂化体的一个重要的贡献结构。与正电荷在碳原子上的另一共振结构相比,这个结构多了一个共价键,这个正电荷是被这个附加的共价键所补偿的。

27
共振论的基本论点 在大多数情况下,能对一个分子写出几个刘易斯结构来描述电子的离域,电子的这种离域是与其稳定性高于单一定域结构的事实有关。这种结构与稳定性的关系或许并不总是正确的,因为在有些分子和离子中,电子离域的结果显然使能量比定域模型有所增加。此外,用两种质体之间共振结构数目的比较作为评价相对稳定性的方法,常常会造成错误,故不作推荐

28
1.1.3 共振规则 共振杂化体是关于含有非定域键的分子的真实结构的一种表示法,是画出数个可能结构,并假定真实分子为它们的共振杂化体。
这些极限式是不存在的仅存在于纸面上,它们是人们的设想。分子不是在它们之间的迅速转变,也不是某些分子具有一个极限式,而另外一些分子具有另一个极限式。 物质的全部分子具有相同的结构,这个结构在所有时间都是一样的,并且为所有极限式的加权平均值。

29
画极限的指导规则 1. 共振结构的原子核的位置必须相同 2. 共振结构必须符合价键的规则
1. 共振结构的原子核的位置必须相同 2. 共振结构必须符合价键的规则 例如,在任何极限式中碳原子不可能为五价,氧原子不可能为三价,围绕氢的价电子数不得超过2,第一行八元素周期的元素最多只能有8个价电子。

30
画极限的指导规则 3. 参与共振的所有原子,即被非定域电子所涉及的原子,必须位于同一平面或近似同一平面。
例如,丙烯基正离子采取平面的几何形状,因为平面结构能使三个p轨道取得最大的重叠。 4. 共振结构必须具有相同数目的未配对电子 如果两个未配对电子的自旋相同,则(II)式为双游离基,(II)式与(I)式所含有的未配对电子数目不同,因此(II)式不是乙烯的共振结构。
式为双游离基,(II)式与(I)式所含有的未配对电子数目不同,因此(II)式不是乙烯的共振结构。")
31
画极限的指导规则 5. 共振结构的能量应彼此大致相同
必须强调一点,共振杂化体为一个单一的物质,不是几个极限式的混合物,共振结构并不代表独立的各个分子。 一种分子只有一种单一的结构。 共振结构的这种描述方法是把各个共振结构综合在一起来描述真实分子。

32
1.2 键能、键长、和偶极 1.2.1键长 在不同分子中键长接近于常数,与取代基所产生的影响几乎无关。碳的键长决定该碳原子的杂化状态。
1.2 键能、键长、和偶极 1.2.1键长 在不同分子中键长接近于常数,与取代基所产生的影响几乎无关。碳的键长决定该碳原子的杂化状态。 表1.2 键长(Å)
")
33
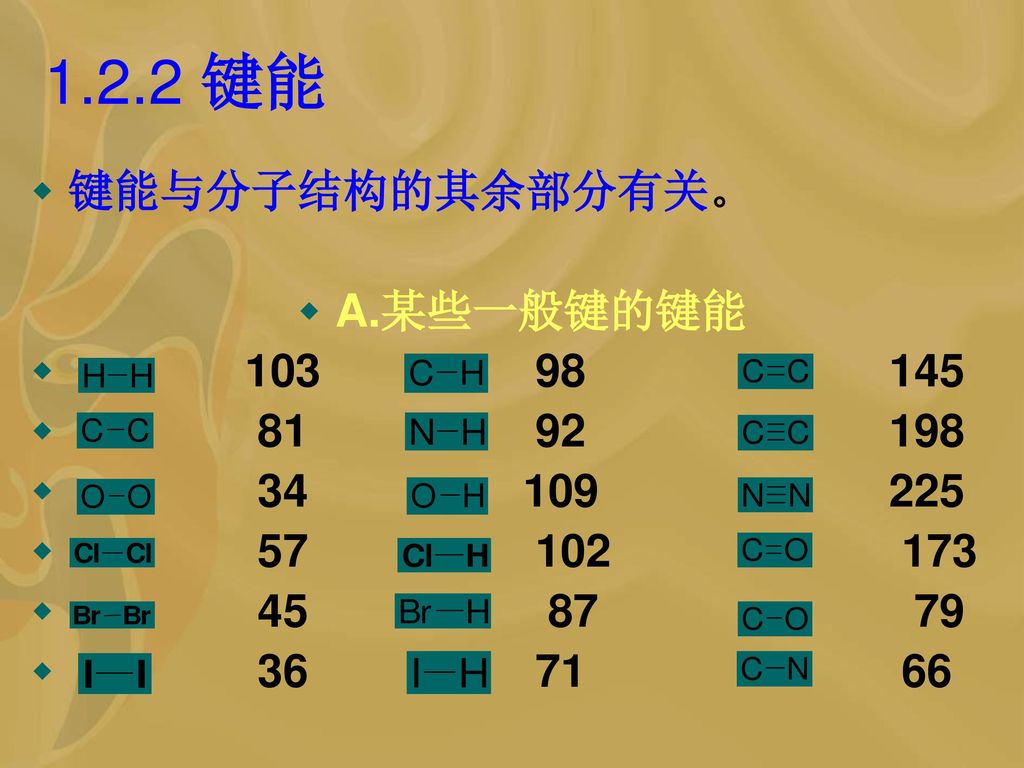
1.2.2 键能 键能与分子结构的其余部分有关。 A.某些一般键的键能 103 98 145 81 92 198 34 109 225
34
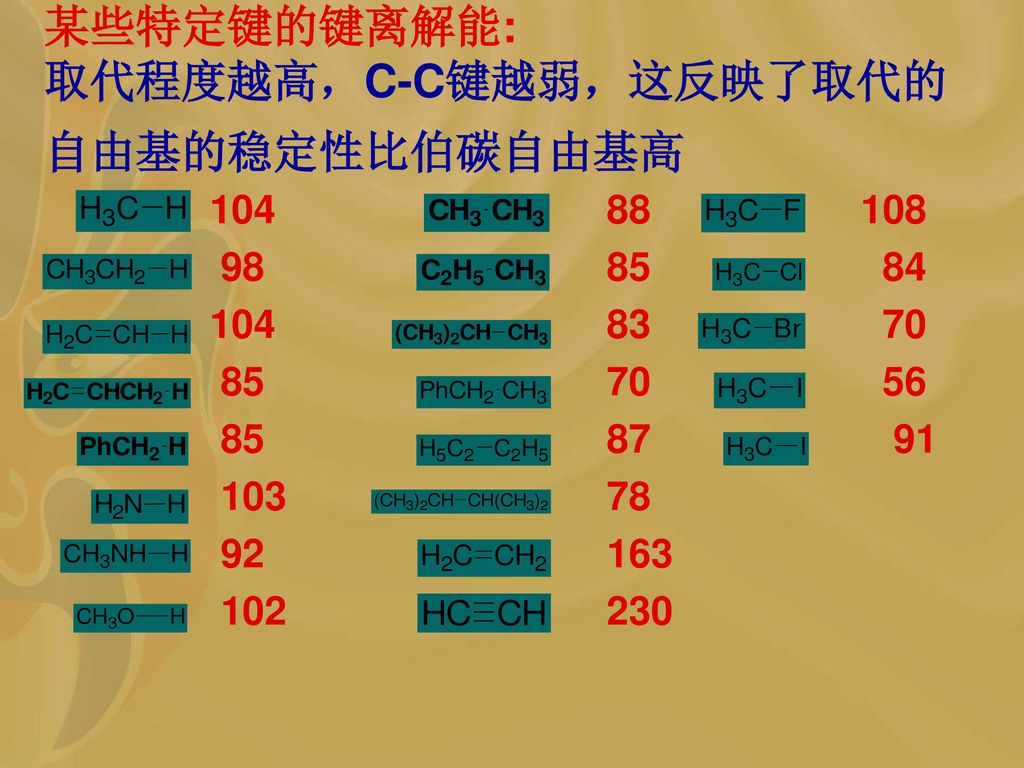
某些特定键的键离解能: 取代程度越高,C-C键越弱,这反映了取代的自由基的稳定性比伯碳自由基高
35
1.2.3生成热 表示标准状态下化合物由组成它的元素生成时所放出的热能。
生成热可以用来精确地比较异构化合物(含有相同的组成元素、每种元素的原子数相同的化合物)的稳定性。 生成热越负,稳定性越高。直接比较元素组成不同的化合物是没有意义的,因为它们生成的键的种类和总数不同。
的稳定性。 生成热越负,稳定性越高。直接比较元素组成不同的化合物是没有意义的,因为它们生成的键的种类和总数不同。")
36
表1.4 一些烃的标准生成热(千卡/摩尔) A.饱和烃 C4 C8 正丁烷 -30.15 正辛烷 -49.82
正丁烷 正辛烷 异丁烷 甲基庚烷 C 甲基庚烷 正戊烷 甲基庚烷 异戊烷 ,2-二甲基己烷 新戊烷 ,3-二甲基己烷 C6 ,4-二甲基己烷 正己烷 ,3-二甲基己烷 2-甲基戊烷 ,2,3-三甲基戊烷 3-甲基戊烷 ,2,4-三甲基戊烷

37
B.烯烃 C4 C6 1-丁烯 -0.03 1-己烯 -9.96 顺式-2-丁烯 -1.67 反式-2-己烯 -12.56
1-丁烯 己烯 顺式-2-丁烯 反式-2-己烯 反式-2-丁烯 顺式-2-己烯 2-甲基丙烯 反式-3-己烯 顺式-3-己烯 C5 甲基-1-戊烯 1-戊烯 甲基-1-戊烯 顺式-2-戊烯 甲基-1-戊烯 反式-2-戊烯 甲基-2-戊烯 2-甲基-1-丁烯 甲基-2-戊烯 3-甲基-1-丁烯 ,3-二甲基-1-丁烯 2-甲基-2-丁烯 ,3-二甲基-1-丁烯 2,3-二甲基-2-丁烯

38
生成热 对于烷烃支链比直链稳定; 对于烯烃双键取代的程度越高,化合物越稳定。
反式烯烃通常比顺式烯烃稳定,这是因为顺式异构体中非成键排斥作用增加的缘故。顺式和反式烯烃氢化结果得到相同的饱和产物,所以它们两个的氢化热的差值就相当于这两个化合物之间的能量差值。

39
1.2.4化学键的极性 共价键中电负性大的元素吸引电子能力强,使共价键中电子的几率分布偏向于电负性较大的原子。
这种电子密度不均衡分布结果产生了键偶极,其单位为电荷与距离的乘积。 具有明显键偶极的键常叫做极性键。 共价键的极性是有机化学中许多结构-反应活性关系的基础。

40
表1.8 一些取代乙酸的pKa值 Cl3CCO2H 0.65 HOCH2CO2H 3.83
Cl2CHCO2H CH3CO2H FCH2CO2H CH3CH2CO2H ClCH2CO2H 电负性较大的原子或基团取代了氢使离子化的平衡常数增加。 电负性大的氟原子使酸性增加的程度比电负性较小的氯原子为大。

41
离解作用使羧酸根带有负电荷,并增加了羧基碳附近的电子密度。
吸电子能力较大的原子使电子的离域程度增大,因此,使取代乙酸离解的平衡常数比乙酸的大。 在丙酸与乙酸比较时,观察到使酸性稍有减弱的效应。 实际上,熵对于取代乙酸离子化作用的影响比焓更为重要。在气相中酸性的测量证实丙酸实际上是一个较乙酸为强的酸,并指出:在水溶液中观察到的相反的情况可能反映了乙酸根离子比丙酸根离子有较高的溶剂化能量。

42
1.3 分子轨道理论 分子轨道理论抛弃了分子中成键电子对定域在特定原子间的概念,而是把电子描述成分布在能量不连续的一系列分子轨道之间。
这个理论以薛定谔方程为基础, Hψ= Eψ ψ是描述轨道的波函数,H是哈密顿算符,E是特定轨道上电子的能量。总的电子能量是各个电子能量的总和。

43
分子轨道理论 在数学上把分子轨道处理为原子轨道的线性组合,因此,波函数ψ是用各个原子轨道剩上权重因子后的总和来表示:
ψ=C1φ1+ C2φ2+…+Cnφn 这种方法称为原子轨道线性组合-分子轨道(LCAO-MO)近似法。所选的原子轨道的这种组合叫做基组集合。 分子轨道的数目(成键的+非键的+反键的)等于产生分子轨道的基组集合中原子轨道的总数:即有多少原子轨道就产生多少分子轨道。
近似法。所选的原子轨道的这种组合叫做基组集合。 分子轨道的数目(成键的+非键的+反键的)等于产生分子轨道的基组集合中原子轨道的总数:即有多少原子轨道就产生多少分子轨道。")
44
分子轨道理论 成键组合的特点是正重叠,其系数的符号相同,而反键组合的特点是负重叠,系数符号相反。见表1.9(P14)
电子填充到轨道中去要遵守填充原理(先占据最低能量的轨道,并且每个轨道最多容纳两个电子)。 根据组成分子轨道(MO)的各原子轨道(AO)的系数和它填充的电子数,可计算出组成它的各原子的电子密度。
。 根据组成分子轨道(MO)的各原子轨道(AO)的系数和它填充的电子数,可计算出组成它的各原子的电子密度。")
45
1.4 休克尔(Hückel)分子轨道理论 休克尔(Hückel)分子轨道理论主要用于处理共轭体系,它认为:π体系能在不考虑共轭的σ骨架的情况下单独地进行处理,并且在决定芳香和多烯化合物的物理、化学和波谱性质时,最重要的正是π体系。由于σ体系和π体系的正交性,所以把它们彼此独立地进行处理是合理的。 平面形共轭体系的σ骨架处于π体系的节面上,所以不与它相互作用。

46
1.4.1 对于线性多烯 它们的分子轨道能级表示为:E=α + mjβ mj=2cosjπ/(n+1) j = 1,2,…,n
当n=2时,即为孤立双键;成键π轨道(j = 1)的能级为:E=α + mjβ=α +2cosjπ/(n+1)β=α +2cosπ/3=α +β;两个成键π电子的总能量为2α +2β。
的能级为:E=α + mjβ=α +2cosjπ/(n+1)β=α +2cosπ/3=α +β;两个成键π电子的总能量为2α +2β。")
47
1.4.1 对于线性多烯 当n=6时,如1,3,5-己三烯,结果见表1.10 (P22):该分子的六个π电子占据ψ1,ψ2,ψ3三个成键轨道,计算出π电子的总能量为α β;这个能量与假定这六个电子分别成对占据三个孤立双键的总能量6(α +β)低0.988β,由此可以看出在己三烯中电子是离域的。
:该分子的六个π电子占据ψ1,ψ2,ψ3三个成键轨道,计算出π电子的总能量为α β;这个能量与假定这六个电子分别成对占据三个孤立双键的总能量6(α +β)低0.988β,由此可以看出在己三烯中电子是离域的。")
48
crj=[2/(n+1)]1/2[sinrjπ/(n+1)]
离域能:对离域电子体系计算出来的电子能量和对交替单键双键计算出来的电子能量之间的差值,称为离域能。 与含定域键的分子相比,含有离域电子的分子提供了额外的稳定性,离域能就是这种额外稳定性的量度。 某碳原子r的2p-AO对第j个MO的贡献相对应的系数,可用下式求出: crj=[2/(n+1)]1/2[sinrjπ/(n+1)]
![crj=[2/(n+1)]1/2[sinrjπ/(n+1)]](http://slidesplayer.com/slide/11612687/62/images/48/crj%3D%5B2%2F%28n%2B1%29%5D1%2F2%5Bsinrj%CF%80%2F%28n%2B1%29%5D.jpg "离域能:对离域电子体系计算出来的电子能量和对交替单键双键计算出来的电子能量之间的差值,称为离域能。 与含定域键的分子相比,含有离域电子的分子提供了额外的稳定性,离域能就是这种额外稳定性的量度。 某碳原子r的2p-AO对第j个MO的贡献相对应的系数,可用下式求出: crj=[2/(n+1)]1/2[sinrjπ/(n+1)]")
49
对组成分子轨道的各原子轨道系数的考察,我们可以看到(如表1
对组成分子轨道的各原子轨道系数的考察,我们可以看到(如表1.10):在最低能量轨道ψ1中全部系数都具有相同的符号,并且在波函数中发生符号改变的次数随轨道能量的增加而增加。 在相邻原子上原子轨道系数的符号改变相当于二者之间的反键相互作用,并且在它们之间的某一点上有一个节面。因此,ψ1没有节面,ψ2有一个,ψ3有两个,到ψ6就有5个节面,且它的分子轨道组合中没有成键相互作用(见图1.13,P22)。 对于成键轨道来说,成键相互作用(正重叠)数目比反键相互作用(负重叠)数目多,而反键轨道的情况正好相反。
:在最低能量轨道ψ1中全部系数都具有相同的符号,并且在波函数中发生符号改变的次数随轨道能量的增加而增加。 在相邻原子上原子轨道系数的符号改变相当于二者之间的反键相互作用,并且在它们之间的某一点上有一个节面。因此,ψ1没有节面,ψ2有一个,ψ3有两个,到ψ6就有5个节面,且它的分子轨道组合中没有成键相互作用(见图1.13,P22)。 对于成键轨道来说,成键相互作用(正重叠)数目比反键相互作用(负重叠)数目多,而反键轨道的情况正好相反。")
50
1.4.2对于环状多烯 它们的分子轨道能级也表示为:E=α+ mjβ
只是mj=2cos 2jπ/n j = 0,±1, ±2,…,{±(n-1)/2,n为奇数时;±n/2,n为偶数时。 n是环中碳原子的数目。 休克尔规则:如果一个质体是由排成平面单环形的原子组成,而每个原子为π体系贡献一个p轨道,并且在那个π体系中电子的总数等于4n+2个(n是整数),那么这个质体就是芳香性的。
/2,n为奇数时;±n/2,n为偶数时。 n是环中碳原子的数目。 休克尔规则:如果一个质体是由排成平面单环形的原子组成,而每个原子为π体系贡献一个p轨道,并且在那个π体系中电子的总数等于4n+2个(n是整数),那么这个质体就是芳香性的。")
51
图中苯的π电子总能量为6α+8β,相当于离域能为2β。

52
弗洛斯特圆圈法 对于含有n个碳原子的环状多烯,就画一个有n个边的正多边形在一个直径为4β的圆圈中,使这个多边形的一个角处于最低点,那么多边形的角接触圆圈的各点就规定了能量。

53
第二章 立 体 化 学 异构体:分子式相同而构造(即化合物中键的特定结合方式和原子的顺序)不同的各种分子集合体之间互称为异构体。包括:碳链异构,官能团异构,官能团位置异构和互变异构。 写出分子式为C5H10的各种异构体 构造相同而原子或基团的空间排列上不同的化合物是立体异构体。包括:构型异构和构象异构。

54
2.1 对映异构关系 手性(chirality)是用来描述一种物体和它的镜象不能重叠时一个名称,历史上手征性和旋光性非常密切相关,化学家们能互换地使用旋光的和手征的这两种描述方法。 旋光性是手征分子的一种性质,即旋转偏振光平面的能力。旋光度的符号和大小都与测量条件有关,包含温度、溶剂和射入样品中的入射光的波长。 一个旋光性的分子一定是手征的,反之亦然。但在一些测量条件下,有些手征分子的旋光性可能非常小,以至无法与零相区别。在这种情况下,可以在另一个波长上测定,得出较大的旋光度。

55
手征中心 碳手征中心: 连在sp3碳上的配基按四面体方式取向,当具有四个不相同的配基时,分子是手征的,我们把这个碳原子称为手征中心。如2-丁醇是手征的,而乙醇却不是。不是手征的分子叫做非手征的。

56
手征中心 除了碳手征中心外,还有硫、磷、氮手征中心,如
具有三个不同取代基的胺原则上是能够有旋光性的因为它们是手征的,但是角锥体翻转所需要的活化能太小,以致不能将对映异构体分离。当它变为季胺盐时,就不能翻转而变成手性的。

57
手性的描述 手性可以用菲舍尔(Fischer)惯用法(D,L)或凯恩-英格尔德-泼莱劳格(Cahn-Ingold-Prelog)惯用法(R,S)来描述 。 注意:分子的旋光度符号和构型(D,L)之间没有简单的关系。不是都象甘油醛那样D-是右旋,L-是左旋;例如,L-丙氨酸这个氨基酸是右旋的。
惯用法(D,L)或凯恩-英格尔德-泼莱劳格(Cahn-Ingold-Prelog)惯用法(R,S)来描述 。 注意:分子的旋光度符号和构型(D,L)之间没有简单的关系。不是都象甘油醛那样D-是右旋,L-是左旋;例如,L-丙氨酸这个氨基酸是右旋的。")
58
手性的描述 因此,D,L与R,S以及旋光度的左旋,右旋它们彼此之间没有必然联系。

59
不含手性中心的手性分子 手性轴和手性面的化合物,如某些旋光性的丙二烯、联芳基、亚烷基环己烷就是轴型非对称分子的例子(手征轴)。反式环烯可作为分子中有非对称性面的例子。
。反式环烯可作为分子中有非对称性面的例子。")
60
手性因素与手性化合物的关系 含有手性中心等手性因素的化合物不一定是手性化合物:如果该化合物含有对称中心,对称面等对称因素则是非手性的。
1, 2-二甲基环丁烷的三个可能的立体异构体是:

61
立体异构体的数目 手征中心的数目和立体异构体的数目之间不存在一个简单的一般关系,因为某些式子可以和它们的镜象重叠。当不存在这些情况时,具有n个不同的不对称中心的体系可能有的立体异构体的数目是2n个。

62
2.2 非对映异构关系 2.2.1 非对映异构体 它们之间不是物体与其镜象的关系的立体异构体。

63
非对映异构体的性质 非对映异构体在化学和物理性质上可以是不相同的。它们具有不同的熔点、沸点、折射率、溶解特性、偶极矩、比旋光度等等。
在非对映异构体中可以把每个手征中心按顺序规则规定为R或S。

64
2.2.2拆分和动力学拆分 拆分是把一个含有等量对映体的混合物(称为消旋体或外消旋混合物)分离成其组分的过程。
用一个旋光性试剂(拆分剂)来处理对映异构体混合物,使其转变为非对映异构体混合物,然后利用非对映异构体的沸点、溶解度等性质的差异,通过蒸馏或重结晶而使其分离,
来处理对映异构体混合物,使其转变为非对映异构体混合物,然后利用非对映异构体的沸点、溶解度等性质的差异,通过蒸馏或重结晶而使其分离,")
65
2-苯基-3-甲基丁酸的拆分为例

66
动力学拆分 用来描述利用与旋光性试剂的选择性反应进行对映异构体分离的一个术语。
它依靠对映异构体与手征试剂反应时速度的差别。一种手征分子与另一种手征分子反应的过渡态能量,对于每一种对映异构体来说都可以是不同的。 如果一个外消旋混合物(R-分子+S-分子)与一个旋光性试剂(R-试剂)反应,那么两个过渡态(R-分子…R试剂和S-分子…R试剂)彼此具有非对映异构的关系。
与一个旋光性试剂(R-试剂)反应,那么两个过渡态(R-分子…R试剂和S-分子…R试剂)彼此具有非对映异构的关系。")
67
动力学拆分

68
酶拆分也是动力学拆分的一类

69
*常用的拆分方法: 晶体机械分离法 形成和分离非对映立体异构体 微生物或酶作用拆分法 色谱分离法 等等

70
(一)晶体机械分离法 原理: 某些外消旋混合物中的(+)-和(-)-对映体自发的一宏观晶体分别析出。当这些晶体键的去便可被看出来时,那么在放大镜的帮助下借助镊子之类的工具来分离。 适用范围:外消旋混合物,且两对映体晶体的区别可被看出来 优缺点:过于繁琐,且不能应用于外消旋化合物和外消旋固体溶液

71
(改良后)接种结晶拆分法: 原理:在外消旋混合物饱和溶液中,用两对映体之一的晶体或其他旋光性化合物晶体小心接种并适当冷却,则其中一种对映体的晶体便会结晶析出 适用范围:外消旋混合物 优点:将该法与重结晶等其他方法合用可获得较好的分离效果

72
例:DL-氯霉素的母体氨基醇的拆分 10g DL-氨基醇和1g D-氨基醇溶解于100ml 80’C水中,冷却至20度后析出D-氨基醇1.9g,然后,将母液在加热至80’C,并加少量水让体积保持100ml,再溶解2g DL-氨基醇于其中,冷却至20‘C,析出氨基醇2.1g,分批连续地溶解DL-氨基醇,可交替地分离出D-氨基醇和L-氨基醇。

73
(二)形成和分离对映立体异构体的拆分 原理:当对映体的酸或碱(+)-A和(-)-A分别和旋光性的碱或酸(-)-B作用后,形成非对映体的两种盐(+)-A(-)-B和(-)-A(-)-B。由于两种非对映体的盐的溶解呈现出颇为明显的差别,于是可以用结晶-重结晶的方法来分离,然后将得到的晶体分解从而得到所需的物质 适用范围:外消旋混合物,外消旋化合物,外消旋固体溶液
-A和(-)-A分别和旋光性的碱或酸(-)-B作用后,形成非对映体的两种盐(+)-A(-)-B和(-)-A(-)-B。由于两种非对映体的盐的溶解呈现出颇为明显的差别,于是可以用结晶-重结晶的方法来分离,然后将得到的晶体分解从而得到所需的物质. 适用范围:外消旋混合物,外消旋化合物,外消旋固体溶液.")
74
**拆分剂所必须具备的几个条件 1. 拆分剂和被拆分的物质的化合物必须容易形成,且又容易被分解成原来的组分
2. 所形成的非对映立体异构体,至少二者之一必须能形成好的晶体,并且两个非对映异构体在溶解度上有可观的差别。 3. 拆分剂应尽量达到旋光纯态 4. 拆分剂必须是廉价的或容易制备的,或在拆分完成之后,能够容易地和接近于定量地回收

75
*实例分析: 外消旋乳酸的拆分: DL-乳酸+吗啡碱(摩尔比 1 :1) H2 O 加热,溶解,冷却
[α]D-91.8度,Yd 95% [α]D-92.7度 (晶体,滤出) (母液) 1. 重结晶 2. 氨水 氨水 吗啡碱 D-乳酸胺 吗啡碱 L-乳酸胺 (回收) 脱色 2. CaCl2 (回收) D-乳酸钙 草酸 L(-)-乳酸锌 D(-)-乳酸 [α ] D-6.83度 m.p ’C , [α]D-2.67度 Yd 50%
 (母液) 1. 重结晶 2. 氨水 氨水. 吗啡碱 D-乳酸胺 吗啡碱 L-乳酸胺. (回收) 1. 脱色 2. CaCl2 (回收) D-乳酸钙. 草酸 L(-)-乳酸锌. D(-)-乳酸 [α ] D-6.83度. m.p. 25.8’C , [α]D-2.67度 Yd 50%")
76
(三)微生物或酶作用下的拆分 原理: 利用酶是专一性的手性催化剂来使其中一个对映体分解从而只剩消另一个异构体达到分离的目的。
原理: 利用酶是专一性的手性催化剂来使其中一个对映体分解从而只剩消另一个异构体达到分离的目的。 一般采用的酶使猪肾酰化酶I,它可催化除天门冬氨酸外的所有普通氨基酸的N-乙酰化衍生物的不对称水解;而对于天门冬氨酸必须用猪肾酰化酶II才能奏效。 这些酶的缺点之一是很不稳定,在使用时要制备新鲜的制剂才能起作用。

77
例: ( N-乙酰-DL-丙氨酸 ) 猪肾酰化酶I 1.乙酸乙酯提取 2.非酶促的酸水解 (在母液中,用离 子交换法分离提纯)
 猪肾酰化酶I 1.乙酸乙酯提取 2.非酶促的酸水解 (在母液中,用离 子交换法分离提纯)")
78
(四)色谱分离法的拆分 A. 柱色谱拆分法 : 吸附剂:用手性的化合物,如淀粉、蔗糖粉、乳糖粉等。
非对称的吸附剂与一个被拆分的外消旋体中的-(+)-分子和-(-)-分子, 分别地形成存在稳定性差别的、具有非对映立体异构关系的两种吸附物,其中之一被吸附得比较牢固,而另一个比较松弛,因此在洗提得过程中,后者比较容易通过吸附剂柱而先被洗脱,于是可以达到拆分外消旋体的目的。
-分子和-(-)-分子, 分别地形成存在稳定性差别的、具有非对映立体异构关系的两种吸附物,其中之一被吸附得比较牢固,而另一个比较松弛,因此在洗提得过程中,后者比较容易通过吸附剂柱而先被洗脱,于是可以达到拆分外消旋体的目的。")
79
B. 配位竞争拆分法: 使含旋光性氨基酸残基的非对称离子交换树脂(固定的配位体)与 或 离子配位,即生成所谓的“配位体交换树脂”。这种配位体交换树脂的固定配位体可以部分地和有倾向地与D-或L-α氨基酸(活动的配位体)发生配位体交换作用,于是可以用来拆分DL-α-氨基酸。
与 或 离子配位,即生成所谓的 配位体交换树脂 。这种配位体交换树脂的固定配位体可以部分地和有倾向地与D-或L-α氨基酸(活动的配位体)发生配位体交换作用,于是可以用来拆分DL-α-氨基酸。 .")
80
C. 纸色谱拆分法: 纤维素具有手性的结构,因此纸色谱分离法也可以被用于外消旋体的拆分。 D. 气相色谱拆分法: 气相色谱拆分法和普通的柱色谱拆分法在基本原理上没有大的差别,它们的不同主要是:在气相色谱法中,用气体(氢气、氦气等)作为携带体,进行气化的被拆分物的扩散和洗脱,而在一般的色谱法中,用液体(各种溶剂)作为被拆分物的携带体。
作为携带体,进行气化的被拆分物的扩散和洗脱,而在一般的色谱法中,用液体(各种溶剂)作为被拆分物的携带体。")
81
2.3 动态立体化学 2.3.1立体专一反应和立体选择反应 立体专一反应是指:在相同的反应条件下,由立体异构的起始物得出立体异构的不同产物。
立体选择性反应是指:在特定反应中,单一一种反应物能够形成两种或更多种立体异构产物,但观察到的是其中一种异构体的形成占优势。

82
某些立体专一反应 对烯的立体专一加成反应:如环氧化反应 亲核取代反应

83
某些立体专一反应 消除反应:如脱卤化氢反应 氧化胺的热解反应

84
某些立体专一反应 在这些立体专一过程中,起始物都是一对对立体异构体,产物彼此也是立体异构体。
每个反应进行时,只得到单一一种立体异构体,而不混杂有另一种立体异构体。

85
某些立体选择性反应

86
构型保留:在反应中立体化学方面有意义的原子的空间排列在反应物和产物中是相同的。
构型转变:在反应中立体化学方面有意义的原子的空间排列在反应物和产物中是对映异构的关系。 如果在生成产物过程中反应物失去了立体化学的完整性,那么这个反应进行时发生外消旋化作用。 如果仅仅丧失一部分立体化学完整性的许多反应实例,我们把它称为部分外消旋化作用。

87
有机分子发生外消旋化作用时最常见的机理过程是:从不对称碳原子处断裂掉一个配基而得出一个平面的三配位中间体。
当不存在象非对称溶剂化作用这样特殊的效应时,这样一个中间体能以两边相等的几率被俘获,结果产生等量的对映异构体产物。 外消旋化反应也可以通过角锥体的翻转作用和围绕碳-碳单键旋转的过程而发生。

88
2.4 潜手征关系 2.4.1同位配基 如果在反应过程中C(2)上的一个质子被另一个配基(例如氘)所取代,则两种可能的取代方式产生同一种产物。所以C(2)上的两个质子在拓扑学上和在化学上都是相当的,它们被称为同位配基。
上的一个质子被另一个配基(例如氘)所取代,则两种可能的取代方式产生同一种产物。所以C(2)上的两个质子在拓扑学上和在化学上都是相当的,它们被称为同位配基。 .")
89
2.4.2 异位配基 如果所发生的过程涉及到C(1)上的两个质子,则结果应得到立体化学上不同的状态。氘取代C(1)上的质子产生一个手征性产物1-氘-1,3-丙二醇:
上的两个质子,则结果应得到立体化学上不同的状态。氘取代C(1)上的质子产生一个手征性产物1-氘-1,3-丙二醇:")
90
C(1)上的两个质子在拓扑学上是不相当的,因为一个氢被取代产生的产物与另一个氢被取代而产生的产物是不同的,它们是立体异构体。这种类型的配基称为异位的,(heteortopic),并且由于取代的产物是对映异构体的(enantiotopic).1,3-丙二醇的C(1)和C(3)都是潜手征中心(prochiralcenters). 如果一个点配基被一个新的点配基取代时产生一个手征组合体(chiral assembly),则原来的组合体就是潜手征的。
,则原来的组合体就是潜手征的。")
91
用字母pro-R 和 pro-S来规定手征分子中的异位配基
选取潜手征中心上的一个异位配基,并且认为它比另一个异位配基有较高的序位,而不打乱其余配基的序位。如果应用顺序规则的结果是:潜手征中心的构型规定为R,则所选的配基是pro-R.如果这个潜手征中心是S,则所选配基是pro-S。 对映异构位的原子或基因,除了对手征试剂的作用以外,在一切化学性质方面都是等当的。

92
2.4.3潜手征面 乙醛羰基有两个面呈现在进攻试剂面前,这两个面彼此具有镜象关系,并且是对应异构位的。
如果从某一个面方向看过去,取代基按递降序位的排列顺序是顺时针的,那么这个面就是re,如果是反时针的,那就是si。

93
一个非手征试剂与一个呈现对应异构位面的分子反应时,将产生等量的对映异构体,得到无旋光性的产物。
用一个手征试剂在潜手征面之间加以辨认是可能的,结果能产生旋光性的产物。

94
非对应异构位 如果一个分子中名义上相当的两个配基被一个试探基团取代后产生的分子是非对映异构的,那么这些配基就是非对应异构位的。
类似地,如果进攻三角式原子的一面产生的分子与进攻另一面所产生的分子是非对映异构体,那么这两个面就是非对应异构位面。

95
非对应异构位 非对应异构位配基的一个重要性质是:它们对于非手征试剂和手征试剂在化学上都是不等当的,并且它们能用物理测试法来加以区别,尤其是NMR波谱法用得最多。



 6 的因数( ) 10 的因数 ( ) 12 的因数 ( ) 14 的因数 ( ) 11 的因数 ( ) 4 的因数( ) 9 的因数( ) 8 的因数( ) 7 的因数( ) 1 、 2 、 3 、 4 、 6 、 12 1 、 11 1 、 2 、 5 、 10.>")















 复习 三角形.>")

.>")